This course provides comprehensive clinical education on tobacco smoke in primary care and public health. It addresses core competencies as well as knowledge, assessment, and treatment-based competencies of healthcare providers. It covers the history of tobacco, epidemiology of tobacco use, tobacco smoke metabolism, dependence, treatment and relapse. It also addresses complications associated with direct and indirect exposure to tobacco smoke, effects of prenatal exposure, methods of screening for exposure, and brief intervention training. This course includes a review of available screening tools, predisposing genetic factors, associated risk and protective factors, withdrawal symptoms and treatment, lab testing procedures, diagnostic tools, and age and gender issues.
- INTRODUCTION
- DEFINITIONS
- HISTORY OF TOBACCO USE AND RESTRICTION
- PREVALENCE AND ECONOMIC IMPACT OF SMOKING
- TOBACCO AND NICOTINE PRODUCTS
- TOBACCO-RELATED CONCEPTS
- CIGARETTE SMOKE
- ANATOMY AND PHYSIOLOGY OF SMOKE INHALATION
- LEARNING OF BEHAVIOR
- SMOKING DEPENDENCE
- HEALTH COMPLICATIONS RELATED TO SMOKING
- COMORBID CONDITIONS
- FETAL EXPOSURE
- PASSIVE SMOKING EFFECTS ON CHILDREN
- PASSIVE SMOKING EFFECTS ON ADULTS
- MEASURING SECONDHAND SMOKE EXPOSURE
- THIRDHAND SMOKE
- INTERVENTIONS FOR SMOKING CESSATION
- REDUCING TOBACCO SMOKE EXPOSURE
- CONCLUSION
- Works Cited
- Evidence-Based Practice Recommendations Citations
This course is designed for physicians, nurses, and other healthcare professionals who may intervene to stop patients from smoking.
The purpose of this course is to provide physicians, nurses, behavioral health professionals, and other members of the interdisciplinary team with a formal educational opportunity that will address the impact of tobacco smoking and secondhand exposure in public health and disease as well as interventions to promote smoking cessation among their patients.
Upon completion of this course, you should be able to:
- Describe the history of tobacco and its impact on society.
- Define the prevalence and economic impact of tobacco smoke exposure on public health.
- Differentiate between available tobacco products.
- Describe the neurophysiologic effects and addictive components of tobacco smoke.
- Describe the anatomy and physiology of smoke inhalation, and outline key points in learning of behavior.
- Define the psychologic and physiologic aspects of smoking dependence.
- List the common health complications related to smoke exposure.
- Identify the common comorbid conditions of tobacco users.
- Describe the developmental complications related to prenatal exposure to smoke.
- Define the effects of exposure to secondhand smoke for children and adults.
- Identify the methods of detecting and measuring tobacco smoke exposure.
- Define thirdhand smoke.
- Outline the methods of tobacco cessation interventions, including necessary considerations for non-English-proficient patients.
- Define the treatment modalities for tobacco addiction, including pharmacologic options.
- Identify strategies to reduce exposure to tobacco smoke.
Mark S. Gold, MD, DFASAM, DLFAPA, is a teacher of the year, translational researcher, author, mentor, and inventor best known for his work on the brain systems underlying the effects of opiate drugs, cocaine, and food. Dr. Gold was a Professor, Eminent Scholar, Distinguished Professor, Distinguished Alumni Professor, Chairman, and Emeritus Eminent Scholar during his 25 years at the University of Florida. He was a Founding Director of the McKnight Brain Institute and a pioneering neuroscience-addiction researcher funded by the NIH-NIDA-Pharma, whose work helped to de-stigmatize addictions and mainstream addiction education and treatment. He also developed and taught courses and training programs at the University of Florida for undergraduates and medical students.
He is an author and inventor who has published more than 1,000 peer-reviewed scientific articles, 20 text books, popular-general audience books, and physician practice guidelines. Dr. Gold was co-inventor of the use of clonidine in opioid withdrawal and the dopamine hypothesis for cocaine addiction and anhedonia. Both revolutionized how neuroscientists and physicians thought about drugs of abuse, addiction, and the brain. He pioneered the use of clonidine and lofexidine, which became the first non-opioid medication-assisted therapies. His first academic appointment was at Yale University School of Medicine in 1978. Working with Dr. Herb Kleber, he advanced his noradrenergic hyperactivity theory of opioid withdrawal and the use of clonidine and lofexidine to ameliorate these signs and symptoms. During this time, Dr. Gold and Dr. Kleber also worked on rapid detoxification with naloxone and induction on to naltrexone.
Dr. Gold has been awarded many state and national awards for research and service over his long career. He has been awarded major national awards for his neuroscience research including the annual Foundations Fund Prize for the most important research in Psychiatry, the DEA 30 Years of Service Pin (2014), the American Foundation for Addiction Research’s Lifetime Achievement Award (2014), the McGovern Award for Lifetime Achievement (2015) for the most important contributions to the understanding and treatment of addiction, the National Leadership Award (NAATP) from addiction treatment providers for helping understand that addiction is a disease of the brain, the DARE Lifetime Achievement Award for volunteer and prevention efforts, the Silver Anvil from the PR Society of America for anti-drug prevention ads, the PRIDE and DARE awards for his career in research and prevention (2015), and the PATH Foundation’s Lifetime Achievement Award (2016) as one of the “fathers” of addiction medicine and MAT presented to him by President Obama’s White House Drug Czar Michael Botticelli. He was awarded Distinguished Alumni Awards at Yale University, the University of Florida, and Washington University and the Wall of Fame at the University of Florida College of Medicine. Gold was appointed by the University President to two terms as the University’s overall Distinguished Professor, allowing him to mentor students and faculty from every college and institute. The University of Florida College of Medicine’s White Coat Ceremony for new medical students is named in his honor.
Since his retirement as a full-time academic in 2014, Dr. Gold has continued his teaching, mentoring, research, and writing as an Adjunct Professor in the Department of Psychiatry at Washington University and an active member of the Clinical Council at the Washington University School of Medicine’s Public Health Institute. He regularly lectures at medical schools and grand rounds around the country and at international and national scientific meetings on his career and on bench-to-bedside science in eating disorders, psychiatry, obesity, and addictions. He continues on the Faculty at the University of Florida College of Medicine, Department of Psychiatry as an Emeritus Distinguished Professor. He has traveled extensively to help many states develop prevention, education, and treatment approaches to the opioid crisis.
Contributing faculty, Mark S. Gold, MD, DFASAM, DLFAPA, has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
John M. Leonard, MD
Jane C. Norman, RN, MSN, CNE, PhD
Alice Yick Flanagan, PhD, MSW
James Trent, PhD
Randall L. Allen, PharmD
The division planners have disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Sarah Campbell
The Director of Development and Academic Affairs has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
The purpose of NetCE is to provide challenging curricula to assist healthcare professionals to raise their levels of expertise while fulfilling their continuing education requirements, thereby improving the quality of healthcare.
Our contributing faculty members have taken care to ensure that the information and recommendations are accurate and compatible with the standards generally accepted at the time of publication. The publisher disclaims any liability, loss or damage incurred as a consequence, directly or indirectly, of the use and application of any of the contents. Participants are cautioned about the potential risk of using limited knowledge when integrating new techniques into practice.
It is the policy of NetCE not to accept commercial support. Furthermore, commercial interests are prohibited from distributing or providing access to this activity to learners.
Supported browsers for Windows include Microsoft Internet Explorer 9.0 and up, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Supported browsers for Macintosh include Safari, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Other operating systems and browsers that include complete implementations of ECMAScript edition 3 and CSS 2.0 may work, but are not supported. Supported browsers must utilize the TLS encryption protocol v1.1 or v1.2 in order to connect to pages that require a secured HTTPS connection. TLS v1.0 is not supported.
The role of implicit biases on healthcare outcomes has become a concern, as there is some evidence that implicit biases contribute to health disparities, professionals' attitudes toward and interactions with patients, quality of care, diagnoses, and treatment decisions. This may produce differences in help-seeking, diagnoses, and ultimately treatments and interventions. Implicit biases may also unwittingly produce professional behaviors, attitudes, and interactions that reduce patients' trust and comfort with their provider, leading to earlier termination of visits and/or reduced adherence and follow-up. Disadvantaged groups are marginalized in the healthcare system and vulnerable on multiple levels; health professionals' implicit biases can further exacerbate these existing disadvantages.
Interventions or strategies designed to reduce implicit bias may be categorized as change-based or control-based. Change-based interventions focus on reducing or changing cognitive associations underlying implicit biases. These interventions might include challenging stereotypes. Conversely, control-based interventions involve reducing the effects of the implicit bias on the individual's behaviors. These strategies include increasing awareness of biased thoughts and responses. The two types of interventions are not mutually exclusive and may be used synergistically.
#91784: Smoking and Secondhand Smoke
Tobacco smoke exposure is a major cause of the nation's most serious and preventable health problems. This course provides comprehensive clinical education on tobacco smoke in primary care and public health. It addresses core competencies as well as knowledge, assessment, and treatment-based competencies of healthcare providers. It covers the history of tobacco, epidemiology of tobacco use, tobacco smoke metabolism, dependence, treatment, and relapse. It also addresses complications associated with direct and indirect exposure to tobacco smoke, effects of prenatal exposure, methods of screening for exposure, and brief intervention training. This course includes a review of available screening tools, predisposing genetic factors, associated risk and protective factors, withdrawal symptoms and treatment, lab testing procedures, diagnostic tools, and age and gender issues.
A clear understanding of tobacco use and smoking is dependent on a knowledge of the basic underlying concepts associated with addiction [1].
Tolerance: The need for greatly increased amounts of the substance to achieve intoxication (or the desired effect) or a markedly diminished effect with continued use of the same amount of the substance.
Withdrawal: Maladaptive behavioral change, with physiologic and cognitive concomitants, that occurs when blood or tissue concentrations of a substance decline in an individual who had maintained prolonged heavy use of the substance. After developing unpleasant withdrawal symptoms, the person is likely to take the substance to relieve or to avoid those symptoms, typically using the substance throughout the day, beginning soon after awakening.
Substance use disorder: A cluster of cognitive, behavioral, and physiologic symptoms indicating that the individual continues using the substance despite significant substance-related problems. There is also an underlying change in brain circuits that may persist beyond detoxification.
Tobacco was the first export of the New World and was marketed in Europe as a remedy for stress, ulcers, headaches, asthma, and even rheumatism. Tobacco's botanical name, Nicotiana tabacum, is derived from Jean Nicot, a French ambassador to Portugal who, convinced of tobacco's medicinal value, sent the plant's seeds to the royal family in France [2].
Tobacco product use has been discouraged in the United States and abroad for centuries. In 1586 the first recorded tobacco prohibition was issued by Pope Sixtus V, who declared it a sin "for any priest to use tobacco before celebrating or administering communion." In 1604, King James I published A Counterblaste to Tobacco, describing smoking tobacco as, "a custome lothsome to the eye, hatefull to the Nose, harmefull to the braine, [and] dangerous to the Lungs" [3]. Tobacco use and distribution saw further restrictions across the globe in the early 1600s. King James I levied heavy taxes on tobacco, the czar of Russia exiled tobacco users, and the Chinese executed persons caught selling tobacco [4].
However, in contrast to strict regulations found elsewhere in the world, tobacco was brought to the United States as a cash crop. The 1880s saw the invention of an automated cigarette-making machine, which paved the way for cigarettes to become the predominant form of tobacco with the start of World War I. The twentieth century also experienced the first major outcry against tobacco in the United States. Though medical concerns were suggested, the first tobacco prohibition movements in the United States were primarily driven by religious and moral motivations. Groups including religious leaders, the Women's Christian Temperance Union, and the Non-smokers Protective supported efforts for prohibition of tobacco. However, strong public resistance against alcohol prohibition also led to the repeal of tobacco restrictions, and by the 1930s these restrictions had all but vanished [5].
One of the lesser known consequences of World War II was that German smoking research and corresponding social change were not acknowledged by the rest of the world. In the 1930s and early 1940s, Germany conducted an aggressive anti-smoking campaign based on medical research from the 1920s and 1930s, which elucidated the carcinogenic effects of smoking. As part of the German movement aimed to preserve a racial "utopia" of pure, healthy Germans, they banned smoking in the workplace, imposed cigarette taxes, restricted advertising and farming, and implemented programs to eliminate smoking [6,7].
Associations between smoking and cancer were not published in the United States until the 1950s and 1960s. The 1964 publication Smoking and Health: Report of the Advisory Committee to the Surgeon General led to immediate political notice of the tobacco issue and the advent of programs and policies to reduce smoking [8]. Anti-tobacco policies have included taxation on tobacco products, increased insurance premiums, warning labels, public health campaigns, and restrictions on tobacco sales to minors, smoking in public areas, and tobacco marketing. Prior to 1964 there were few if any laws regulating involuntary secondhand smoke (SHS) exposure. Studies revealing the detrimental effects of SHS to nonsmokers led to new anti-smoking legislation. As of June 2009, the General Services Administration (GSA) has established smoke-free environments for federal facilities. Interior areas previously designated for smoking have been closed and smoking is prohibited in courtyards and within 25 feet of doorways and air intake ducts in outdoor spaces [9]. Further, nearly all 50 states have laws restricting smoking in places such as schools, public transportation, government buildings, elevators, and restaurants. In accordance with federal law, smoking is prohibited on buses, trains, and domestic airline flights. Such laws have decreased cigarette consumption by making smoking less socially acceptable and more inconvenient [5].
On June 22, 2009, President Barack Obama signed HR1256: The Family Smoking Prevention and Tobacco Control Act. This was enacted as a result of several findings made by Congress, specifically that almost all new users of tobacco products are younger than the minimum legal age to purchase such products. Under this law, the U.S. Food and Drug Administration (FDA) now has the authority to regulate tobacco products [10]. The FDA had previously attempted to assert jurisdiction under the Food, Drug, and Cosmetic Act in 1996 to regulate tobacco advertising, labeling, and purchasing restrictions (e.g., federal minimum age of 18 years and requiring retailers to check identification). However, the tobacco industry retaliated by suing the federal government, as there was no set legislation to give the FDA this authority. As a result, all FDA regulations were dropped [11]. Due to the 2009 law, the FDA can now establish a minimum age of sale of tobacco products, test and report on tobacco product ingredients/additives, prohibit cigarettes from containing any flavors other than tobacco or menthol, and apply the same restrictions on labeling and advertising of cigarettes to smokeless tobacco products. Of note, this law states that the FDA cannot ban existing products or require nicotine be eliminated from any product.
In 2017, the FDA unveiled a comprehensive plan on tobacco and nicotine regulation to reduce the number of preventable deaths caused by smoking and tobacco use [472,474]. The two key areas of focus of this plan are reducing the nicotine levels in combustible cigarettes to render them minimally or nonaddictive and harnessing new forms of nicotine delivery that could allow currently addicted adult smokers to get access to nicotine without many of the risks associated with using combustible tobacco products. Similar to the 2009 policy, this plan also explores the extent of tobacco flavoring in attracting youth and new smokers; menthol flavoring will be included in this plan. Of note, this policy only affects newly regulated tobacco products and will not affect any current requirements for cigarettes and smokeless tobacco. In 2019, President Donald Trump signed legislation to amend the Food, Drug, and Cosmetic Act to raise the federal minimum age to purchase all tobacco products (including e-cigarettes) from 18 to 21 [475]. It is now illegal to sell tobacco products to anyone younger than 21 years of age.
As of April 2022, there are three companies approved to sell 15 modified-risk tobacco products (MRTPs), including cigarettes, smokeless tobacco (snuff), and a heated tobacco product [476]. To receive a MRTP authorization, the FDA must find that the product is less likely to cause disease, including cancer, cardiovascular disease, emphysema, and bronchitis, than traditional cigarettes and must discern whether those who do not use tobacco products would start using the product and whether existing tobacco users who would have otherwise quit would switch to the modified risk product instead [477].
Approximately 480,000 Americans die each year as a result of active and/or passive smoking-related health consequences [12]. Despite the seemingly well-known and highly publicized health consequences of smoking, 13.9% of the U.S. population 18 years of age or older are current cigarette smokers [460]. Former U.S. Assistant Secretary for Health Howard Koh asserted that although evidence-based tools were successful in substantially reducing smoking prevalence between 1997 and 2004, efforts were not applied to their full potential nationwide, limiting the efficacy of anti-smoking campaigns [14]. Other experts have attributed declines in cigarette smoking to anti-smoking advertisements, stigma, smoking bans, and increased taxation [460]. Evidence-based tools remain valuable, indicated by slow, steady downward prevalence trends since 1997. However, they are only useful if they reach an audience. These tools seem not to be preventing the initiation of new smokers, despite the overall reductions in use [14,15].
Nearly 1.6 million Americans initiated cigarette smoking in 2019, continuing a downward trend noted since 2006 (down from approximately 2.5 million); 34% of these were 12 to 17 years of age [13]. About one-third of new smokers will ultimately die from a smoking-related illness [16]. Higher levels of education are correlated with a lower likelihood of having smoked cigarettes in the past month [13]. The number of first-time cigar users is slowly declining, from 3.4 million in 2006 to 2.1 million in 2019 [13]. In 2019, use in the past year of any tobacco product was highest among American Indians/Alaska Natives (39.8%) followed by persons of two or more races (35.2%), White Americans (28.6%), Black Americans (27.2%), Hispanics (19.5%), and Asians (13.2%) [13].
Approximately 41,000 adult nonsmokers die each year from exposure to SHS, and this continues to be a significant environmental risk in the United States [12]. In a 2009 study, the prevalence of smoking in New York City was lower than the national average (23.3% vs. 29.7%), but the proportion of nonsmoking adults with elevated cotinine levels was higher (56.7% vs. 44.9%), especially among Asians, even nearly two years after implementation of smoke-free workplace legislation [20]. This finding was attributed to the large amounts of people living in close proximity (26,000 people and 10,000 housing units per square mile vs. the national average of 80 people and 33 housing units per square mile) [20]. In a 2017 study, Perlman and colleagues examined cotinine levels in New York City nonsmokers, and found that 37.1% had elevated levels [17]. It is thought that this reduction (from 56.7% in the earlier study) is a result of smoke-free air policies enforced within the previous 10 to 15 years. The researchers also noted that greater population density and pedestrian exposure continued to contribute to a high number of nonsmokers with elevated cotinine levels compared with the national average [17]. Nonsmoking individuals with the highest cotinine serum concentrations tended to be living in high-poverty neighborhoods, have lower educational attainment, be 20 to 39 years of age, report non-Hispanic Black race, and be male.
Tobacco use is one of the most expensive addictive behaviors in the United States. In 2015, an estimated 299.9 billion cigarette stick equivalents of combustible tobacco products (based on the weight of 0.0325 ounces of tobacco per cigarette) were consumed in the United States, of which 267 billion were cigarettes [21]. The Federal Trade Commission (FTC) reported that 203.7 billion cigarettes were sold in the United States in 2020 [54]. Americans spent $84.8 billion on cigarettes alone (10.6 billion packs) in the 2021 fiscal year [23].
Smoking-related costs in the United States are staggering. The total annual public and private healthcare expenditures caused by smoking are estimated to be greater than $300 billion, including nearly $170 billion in direct medical costs and more than $156 billion in lost productivity related to premature death and exposure to SHS [12].
Cigarette smoking is on the decline in the United States, but use of other tobacco products is not [13,21]. In addition to a rise in use of smokeless tobacco, people across the United States (especially youth) are using e-cigarettes, cigars, cigarillos (small cigars), hookahs, kreteks, pipes, and bidis (or beedis) [18,25]. Unfortunately, each of these products is just as dangerous (if not more so) as use of cigarettes. Cigarettes are defined by the U.S. Department of the Treasury as "any roll of tobacco wrapped in paper or in any substance not containing tobacco," while cigars are defined as "any roll of tobacco wrapped in leaf tobacco or in any substance containing tobacco" [26]. Cigars also differ from cigarettes in processing; they consist of filler, a binder, and a wrapper, all made of air-cured and fermented tobaccos [27]. Cigars show significant variability in physical and chemical characteristics, with total nicotine content ranging from 10.1 mg to 444 mg per cigar, length ranging from 68.0 mm to 213.5 mm, and diameter ranging from 8.0 mm to 20.5 mm [28]. Due to their size and makeup, smokers can spend up to an hour smoking a single cigar; therefore, its ensuing effects (e.g., rates of cancer, chronic obstructive pulmonary disease [COPD]) are more pronounced. Cigarillos, or "little cigars," are generally about half the size of a normal cigar, weighing 1.5–3 g on average [29]. Many types are made to look like cigarettes and are sold in packs of 20 with filter tips. Cigarillos are perceived as a less addictive, less harmful, and less expensive alternative to cigarette use [30; 31].
Nicotine—the identified drug in tobacco—is highly addictive, and flavors enhance nicotine's addictive effects. Flavors significantly increase tobacco use because they enhance its appeal, especially among adolescents and young adults [481]. More than 90% of current smokers started smoking as teenagers, and it is estimated that 80% of youths who used tobacco began with flavored tobacco products [481]. Additionally, flavors, such as menthol, mask the harshness and bitterness of tobacco, sustain tobacco dependence, and hinder cessation.
Aggressive and targeted marketing of flavored tobacco products has long been an industry tactic intended to lure young people into experimentation with tobacco products, resulting in addiction and, consequently, premature death. The tobacco industry has especially targeted Black and LGBTQ communities with predatory marketing of menthol cigarettes and flavored cigars. As of 2019, the FDA found there were 18.5 million people in the United States who smoked menthol cigarettes, which are disproportionately used by marginalized populations [481]. Nearly 85% of African American smokers use menthol cigarettes.
An estimated 9,000 Americans die prematurely from cigar smoking each year. Additionally, an annual health care expenditure of $1.8 billion is attributed to cigar use. Flavors are critical to cigar usage. There is a greater frequency of smoking by adults who smoke flavored cigars compared with those who smoke unflavored cigars. In 2020, an estimated 960,000 youths smoked a cigar at least once in 30 days, with almost 60% reporting that they used flavored cigars.
Due to increased federal taxation on cigarettes, cigarette tobacco, and small cigars, many consumers apparently switched to smoking products virtually identical to cigarettes or small cigars, but classified as large cigars, or from smoking cigarette rolling tobacco to smoking "pipe tobacco" [22]. Subsequent to the 2009 tax increase and intensified FDA regulation, many companies simply relabeled cigarette rolling tobaccos as pipe tobaccos (not subject to increased taxation) [21]. Sales of "pipe tobacco" increased from 5.2 million pounds in 2009 to 43.7 million pounds in 2013 (a 740% change) while rolling tobacco sales dropped from 21.3 million pounds to 3.8 million pounds [22]. Following a similar relabeling and marketing effort for small cigars, sales of large cigars jumped from 5.8 billion sticks in 2009 to more than 12.4 billion sticks in 2013, while small cigars decreased from 5.7 billion sticks to 0.7 billion sticks in the same years. In 2016, the FDA extended its limitations for tobacco products to include e-cigarettes, vaporizers, and other electronic nicotine delivery systems [458]. As a result, these products must include warnings and manufacturers must submit documentation to the FDA for review and limit sales to persons 21 years of age or older. The goals of these regulations are to increase public health awareness and, especially, reduce marketing and sales to adolescents and young adults, who are commonly targeted by providing tobacco flavors including apple, cherry, cream, grape, "jazz," strawberry, and wine. Before this ruling, there were no federal laws restricting sales of these types of products, but an alarming increase in unregulated tobacco products, especially among high school students, prompted the FDA to enforce regulations. In 2018, the FDA issued more than 1,300 warnings and fines to retailers who illegally sold e-cigarette products to minors [464].
Prohibiting menthol in cigarettes has been proposed as an approach to decrease the appeal of cigarettes and ease of smoking, thus minimizing the likelihood of smoking initiation and subsequent nicotine dependence. It would also improve the health of current smokers by decreasing cigarette consumption and increase the likelihood of cessation. This would also minimize death and disease associated with exposure to secondhand smoke. The FDA estimates that prohibiting menthol in cigarettes would prevent 654,000 premature deaths in 40 years [481]. The FDA also found prohibiting menthol would advance health equity, because menthol use is more prevalent in marginalized communities, especially among Black smokers, and prohibiting menthol would lessen the health harms those communities disproportionately bear. It is estimated that the menthol prohibition would prevent 238,000 premature Black deaths in 40 years [481]. In April 2022, the FDA proposed a rule banning menthol flavoring added to cigarettes [481].
The FDA also proposed a rule that would prohibit flavors (including menthol) in cigars and their components and parts. As noted, flavors appeal to young people, and cigar flavors come in many varieties (including spice, strawberry, grape, banana, licorice, menthol, and chocolate) that make cigars easier to smoke.
Similar to the proposed ban on menthol in cigarettes, this proposed rule would prohibit the manufacture, distribution, or sale of flavored cigars in the United States. Here, too, the FDA focuses on the supply side of the market and would not prohibit individual consumers from possessing or using flavored cigars.
The proposed rule comprehensively defines cigars—which are made in different sizes and shapes—as "a roll of tobacco wrapped in leaf tobacco or any other substance containing tobacco" [481]. This broad definition captures many tobacco products (including little cigars, cigarillos, and large cigars) and should guard against manufacturers skirting the ban by switching to other products that closely resemble other prohibited flavored tobacco products. The flavor prohibition would also apply to cigar "components or parts," so products such as filters, blunt wraps, or tips also could not be flavored [481].
The rise of e-cigarettes in the past decade has introduced new variables in the prevention and treatment of nicotine addiction. Originally marketed as a smoking cessation tool, e-cigarettes are electronic products that typically deliver nicotine in the form of an aerosol [456]. Most e-cigarettes consist of a cartridge (which holds a liquid solution containing varying amounts of nicotine, flavorings, and other chemicals), a heating device (vaporizer), and a power source (usually a battery) [457]. In many e-cigarettes, puffing activates the battery-powered heating device, which vaporizes the liquid in the cartridge. The resulting aerosol or vapor is then inhaled (called "vaping") [457]. It is unclear if this delivery method decreases the risks seen with conventional tobacco smoking; however, it does introduce the risks of toxicity associated with consumption of the potent e-liquid, including heavy metals (e.g., cadmium, chromium, lead, manganese, nickel) that are also emitted from the heating element and heated plastic [478].
In 2020, 3.7% of adults were current every day or some days e-cigarette users. Adults 18 to 24 years of age (9.4%) have the highest rate of e-cigarette use, followed by those 25 to 44 years of age (5.2%), 45 to 64 years of age (2.2%), and older than 65 (0.6%) [456]. Use is much higher among men (4.6%) than women (2.8%). Current use of e-cigarettes among high school students skyrocketed from 1.5% in 2011 to 11.3% in 2021, making it the number one form of nicotine used among high school-age youth [331,459]. E-cigarette use is particularly prevalent in White students (14.5%) and is less prevalent among Black (5.9%) and Hispanic (7.6%) students. Slightly more high school age girls (11.9%) than boys (10.7%) use e-cigarettes. In 2021, 2.8% of middle school students were current users of e-cigarettes [331].
According to the Centers for Disease Control and Prevention (CDC), large cigar consumption increased 115% from 2000 to 2020, with cigar smoking being the third most common form of tobacco use among youth [32,33,331]. Cigar use is twice as common among Black versus White high school students and is much more common among boys than girls. However, it has been shown that adolescent (and likely adult) cigar use is significantly underestimated due to systematic misreporting on statewide surveys, which is mainly attributed to the language and definitions used in questions that assume knowledge of all types of cigars [34]. For example, it was found that more than half of Black & Mild (brand of cigars and cigarillos) users did not report any cigar/cigarillo use on a 2009 Virginia survey, largely because the usage of the terms "cigar" or "cigarillo" for this (and other similar products) is not common in the youth- or culture-specific lexicon.
Bidis consist of sun-dried tobacco, finely ground and rolled into a leaf of the Diospyros melanoxylon plant native to India. They contain concentrated tobacco, with an average 21.2 mg/g of nicotine compared with 16.3 mg/g of nicotine in filtered and 13.5 mg/g in unfiltered cigarettes, but have less total nicotine because they are shorter [35]. Nonetheless, an unfiltered bidi can release three to five times more tar and nicotine and contain more ammonia and carbon monoxide (CO) than a regular cigarette. Bidis look similar to small cigars or marijuana cigarettes and are available filtered or unfiltered in many flavors, including vanilla, chocolate, strawberry, cherry, and menthol [36]. Bidis are not commonly used in the United States, and sale and distribution is banned in some states (e.g., Illinois, Vermont, West Virginia). However, these products are available on the Internet [37].
Kreteks, or clove cigarettes, are composed of a mixture of tobacco (60% to 80%) and ground clove buds (20% to 40%), available with or without filters [38]. A popular, representative kretek brand contains less nicotine than popular cigarettes (7.39 mg), but smokers extract equal amounts of nicotine by altering smoking behavior [39]. For example, clove cigarettes can be smoked slower, using more puffs. Overall, smokers will do whatever is necessary to achieve plasma levels of nicotine comparable to their usual brand of cigarette.
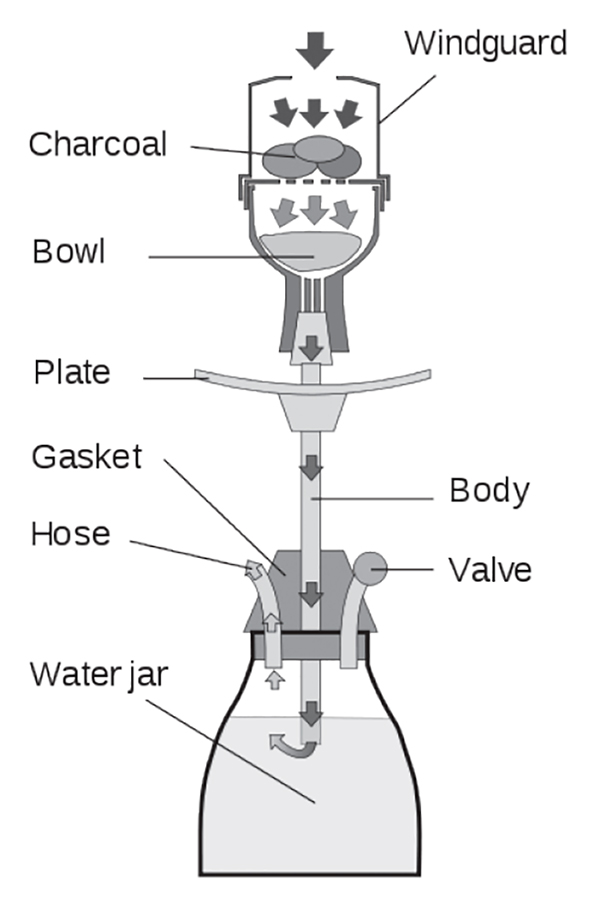
A hookah is a type of waterpipe comprised of a head or bowl, plate, body, jar, hose, and mouthpiece (Figure 1). The body of the hookah fits down into the jar, which is partially filled with water, although any liquid (e.g., alcohol, juice) can be used. Tobacco is placed in the bowl at the head of the body and covered with a filter, such as perforated tin foil, and then burning embers or charcoal is placed above it (and sometimes covered by a cap). The hot air from the charcoal roasts the tobacco and the ensuing smoke is passed down into the liquid in the jar where it is partially filtered, diluted, and cooled. The smoke then bubbles up and passes through the hose and mouthpiece for inhalation. Repeated inhalation is required to keep the tobacco burning. The plate stores dead coals/embers. The types of tobacco used for hookah are ajami or tumbak, which is a pure, dark tobacco paste; "honeyed" or tobamel or maassel, containing 70% honey or molasses and featuring flavors (e.g., apple, mango, banana); or jurak, which may be sweetened or contain fruits or oils. It is commonplace to use 10–20 g at a time, and these tobaccos may be mixed with other drugs [40]. Smoking sessions last up to an hour or longer, and it has been reported that the nicotine content of the tobacco used for hookah is higher than that in cigarettes [41]. Thus, the smoker is exposed to a higher volume of smoke for longer periods (not to mention those in the vicinity). A report from the World Health Organization states that a hookah user may inhale as much smoke in one session as a cigarette smoker would after consuming at least 100 cigarettes [42]. Contrary to popular belief, waterpipe smoking is not safer or less addictive than cigarette smoking [43]. The FDA began regulating the manufacture, import, packaging, labeling, advertising, promotion, sale, and distribution of tobacco mixtures used for hookah in 2016 [24]. Hookah smoke contains higher concentrations of CO, nicotine, tar, heavy metals, and carcinogens, likely because of its method of use (i.e., tobacco mixtures heated by quick-burning charcoal or wood embers and inhalation through use of a plastic hose for an hour or longer) [44,45]. It is also common to share a hookah, so users are also at risk of exposure to infections (e.g., herpes due to sharing of the mouthpiece) [46]. Hookah pipe smoking may be a gateway to cigarette smoking and other drug use. Although policies are in place to ban smoking in many public places, many times, hookah use is exempt because it is done in places which identify themselves as "tobacco bars," waterpipe smoking areas are set up outside, or the smoking is done in places where tobacco is sold.
For many years, efforts to make cigarettes "safer" have been pursued as a compromise solution [48]. Filtering devices have been used to selectively reduce cigarette smoke constituents for almost 60 years [49]. Studies from the 1970s concluded that charcoal filters can remove up to 66% of ciliotoxic agents from mainstream smoke, and cellulose acetate filter tips can eliminate up to 75% of N-nitrosamines, which are known volatile carcinogenic compounds [50,51]. However more recent studies have shown that neither type of filter is effective for reducing the free radical and reactive oxygen species content in the particulate or gas phase of cigarette smoke [52]. Additionally, remnant (i.e., post-filter) aqueous tar can cause the formation of DNA adducts, particularly the mutagenic 8-Oxo-2'-deoxyguanosine (8-oxo-dG).
The FTC performed tar, nicotine, and CO content measurements in all domestic cigarette varieties sold in the United States, which numbered almost 1,300 in 1998, the last year the report was conducted. The FTC defines tar as the particulates of cigarette smoke minus water and alkaloids, such as nicotine, detected using a method developed in 1966 [53]. In 2020, 99.8% of cigarettes sold in the United States had filters, and the FTC reported that in 2016, 87.9% of the market share of cigarettes had less than 15 mg of tar (manufacturer reported), compared with only 2% in 1967 [53,54]. Nevertheless, epidemiologic evidence does not indicate that modern cigarettes are any safer. Smokers participating in the Cancer Prevention Study II (CPS-II) from 1982 to 1988 manifested an almost sixfold increase in lung cancer death compared to Cancer Prevention Study I (CPS-I) participants during 1959 to 1965, even though filter tips were introduced in the 1950s and only the latter group benefited from their implementation [55]. Smoking pattern compensation and use of stronger tobacco strains may be at least partially responsible for this paradoxical trend.
Filter vents, usually shaped in rings of small perforations along the filter, allow air to mix with smoke, diluting the amount of tar, nicotine, and CO detected by the FTC method [53]. Interestingly, as many as 58% of smokers of cigarettes with tar less than approximately 7% (formerly labeled "ultralight") and 53% of smokers of cigarettes with tar levels of 8–14 mg of tar (formerly labeled "light") inadvertently cover these vents to some extent [56,57]. Blocking half of the vents of a 4.4 mg tar cigarette, as is done when smokers pinch the cigarette with their fingers or hold the cigarette in their lips, increases yields of tar by 60%, nicotine by 62%, and CO by 73% [58]. Poor reliability of the FTC method is further made evident in the work of Byrd and Robinson, who concluded that the "FTC yield cannot precisely predict nicotine uptake for an individual smoker" and "nicotine uptake by smokers is influenced by…many possible smoker-controlled parameters" [59]. Interestingly, this publication originates from the R.J. Reynolds Tobacco Company. Another contributing factor to the increase in mortality related to smoking may be the concentration of nitrate in tobacco leaves, one of the most important precursors for the endogenous formation of N-nitrosamines during smoke inhalation [60]. Cigarette nitrate content has increased from 0.5% in the 1950s to 1.2% to 1.5% in the late 1980s, possibly due to the increased use of chemical fertilizers and the introduction of plant ribs and stems into U.S. tobacco blends [61]. The carcinogenic potential of nitrosamines has been well documented.
All in all, efforts to reduce the health hazards of smoking leave much to desire, and in spite of filter tip implementation and reportedly lower tar values, cigarettes remain a serious health hazard, affecting smokers and those around them.
Cigarette smoke is a complex mixture of more than 7,000 components, including nicotine, aromatic hydrocarbons, sterols and oxygenated isoprenoid compounds, aldehydes, nitriles, cyclic ethers, and sulfur compounds [62,63,134]. At least 70 of these components are known to cause cancer [134]. Firsthand smoke is defined as the smoke that the smoker inhales. Smoking tobacco products also generates environmental tobacco smoke, also known as SHS and passive smoke, which consists of both exhaled mainstream and sidestream smoke. These two forms of smoke differ in chemical composition and have different temperatures and oxygen levels during generation. The burning end of a cigarette produces sidestream smoke, which in turn is the main component of SHS. Some known toxins of the thousands of chemical constituents in tobacco smoke are also present in SHS, including benzene, cadmium, ethylbenzene, formaldehyde, hydrazine, lead, limonene, methylamine, methylene chloride, nicotine, pyridine, toluene, and radioactive polonium-210 [64,65,66]. One study identified indoor air pollution from SHS as 10 times greater than diesel car exhaust [67].
Many of the diseases once thought only to be caused by active smoking have now been authoritatively linked to environmental tobacco smoke [62,68]. This finding is not surprising considering that many of the harmful components found in both firsthand smoke and SHS are more concentrated in SHS. Nicotine, tar, nitric oxide, and CO levels have been shown to be nearly twice as concentrated in SHS. Other harmful chemicals preferentially formed in SHS include carcinogenic aromatic amines (e.g., o-toluidine, 2-naphthylamine, and 4-aminobiphenyl) [62,65,69]. Three times greater concentrations of polonium-210 exist in sidestream smoke, because most of the radionuclides are not deposited in the smoker's lungs, as with mainstream smoke [479].
According to Lans et al., the crushed leaves of Nicotiana tabacum are applied to wounds in Guatemala, and tobacco steam vapor is considered a cure-all in Latin America and the Caribbean. In addition to its most addictive component, nicotine, the tobacco plant contains many enzymes, flavonoids, and coumarins and malic, citric, and phenolic acids [70]. In a case-control study by Sandler et al., tobacco use and secondhand exposure (e.g., parents had smoked) reduced the risk of developing ulcerative colitis; however, at least one meta-analysis found that nicotine therapy for existing ulcerative colitis, while better than placebo, was not more effective than standard treatment and was associated with significant adverse events [71,430]. Plants of the genus Nicotiana have been manipulated in various experiments to express proteins that may be used medicinally. Indeed, transgenic tobacco plants have been used in the development of vaccines for measles, lymphoma, and diabetes [72,73,74].
Administration of any drug via smoking is a highly efficient route, allowing rapid delivery to the brain. This act involves inhalation of a small volume of smoke (an average of about 35 mL for cigarettes) into the mouth from which it is drawn into the lungs [75]. The breathing pattern employed is different from normal tidal breathing in that a smoker's inhalation is deeper and more rapid, drawing the smoke in as a bolus at the beginning of inhalation [76]. However, this pattern varies greatly between smokers and during the course of consuming a single cigarette [77]. Uptake of smoke ingredients is determined by many factors, including chemical composition, smoker's inhalation behavior, lung morphology, and physiologic parameters such as tidal volume, vital capacity, rate of breathing, and rate of lung clearance [78]. Individual differences in size, metabolism, and genetics may also play a role. One hypothesis suggests that stimulation of nicotine-sensitive receptors in the upper airway by various elements of smoke governs the amount inhaled. Indeed, application of a topical anesthetic to the upper airway reduces the quantity of smoke inhaled [79].
Tobacco smoke consists of gaseous and particulate phases, with the particulate phase comprising about 8% of the total volume [76]. Particulate deposition depends on the size, shape, and hygroscopicity (ability to absorb water vapor) of the particles as well as the duration and depth of inhalation [77]. Smoke particles range from 0.1–1.0 mm in diameter as they exit a cigarette, doubling in size within half a second due to aggregation, cooling, and condensation [80]. Larger particles (1–5 mm) are likely to deposit in the trachea and bronchi, whereas smaller particles (0.01–1 mm) reach bronchioles, alveolar ducts, and alveoli. Irregularly shaped or fibrous particles tend to get trapped at branching points, although some of these particles can travel on to the alveoli [81]. Interestingly, smoking seems to result in a greater apical and central distribution of particles than normal tidal breathing. This finding may help to explain the pathogenesis of centrilobular emphysema [76].
Cigarettes deliver nicotine in a pulsatile manner, with plasma concentrations reaching their peak within 1.5 to 3 minutes of the commencement of smoking and gradually returning toward baseline within two to three hours [82]. Thus, nicotine levels rise and fall throughout the day with each cigarette smoked, declining to minimum amounts found in nonsmokers in the morning after the extended abstinence period of sleep. Such continuous flux in blood nicotine levels locks the user into an endless cycle of ups and downs and is thought to lead to the commonly held notion that smoking has a positive effect on mood. Considering smokers begin to experience withdrawal symptoms within hours of their last cigarette, and because these unpleasant effects are almost completely alleviated by smoking, this perception is hardly surprising. Daily repetition of this process links these perceived positive health benefits to the act of smoking in the smoker's mind and often results in the false identification of cigarettes as an effective form of self-medication [83].
What is it about smoking that makes it so addictive? On one hand, this form of drug delivery is very efficient; inhaled nicotine is absorbed through pulmonary rather than systemic circulation and can reach the brain within 10 to 20 seconds [84]. Once inside the central nervous system (CNS), nicotine stimulates release of dopamine from the nucleus accumbens, much like the use of cocaine and amphetamines, leading to the feeling of satisfaction and well-being. Given such rapid central reinforcement, it is not surprising that tobacco can become highly addictive. On the other hand, familial and social influences often play a crucial role in determining who might start smoking, quit, or become dependent [83]. For example, one study managed to train a small percentage of rhesus monkeys to smoke, but with such difficulty that it concluded that "environmental factors play the primary role in developing smoking behavior" [85].
The U.S. Preventive Services Task Force recommends that primary care clinicians provide interventions, including education or brief counseling, to prevent initiation of tobacco use among school-aged children and adolescents.
(https://jamanetwork.com/journals/jama/fullarticle/2765009 Last Accessed: May 11, 2022)Level of Evidence: B (There is high certainty that the net benefit is moderate or there is moderate certainty that the net benefit is moderate to substantial.)
Experimenting with smoking usually occurs in the early teen years and is predominantly driven by psychosocial motives [83]. For a first-time user, lighting a cigarette is a symbolic expression of autonomy and independence; acquisition of the desired image is often a sufficient incentive for a novice smoker to tolerate the body's rejection of the first few cigarettes. Despite an admitted awareness of at least some of the deleterious effects of smoking, in 2018, 1 in 4 high school students and 1 in 14 middle school students admitted to using a tobacco product in the past 30 days [135]. Almost all people (90%) who will smoke as adults have started doing so by 18 years of age, and the earlier a person begins, the more likely they are to continue [135]. Within a year, adolescents inhale the same amount of nicotine per cigarette as adults, and they too experience the craving and withdrawal symptoms associated with nicotine addiction [83]. By 20 years of age, 80% of smokers regret ever having started.
Much research has been dedicated to uncovering reasons for the development of a smoking habit. Risk factors include [86]:
Presence of a smoker in the household
Single parent home and/or strained relationship with parent
Comorbid psychiatric disorders
Low level of expressed self-esteem and self-worth
Poor academic performance
In boys, high levels of aggression and rebelliousness
In girls, preoccupation with weight and body image
Increased adolescent perception of parental approval of smoking
Affiliation with smoking peers
Availability of cigarettes
In addition, twin studies revealed a significant genetic contribution to both smoking initiation and dependence [87,88].
In practice, many find the very act of smoking a cigarette ritualistic and calming. The process of "packing" cigarettes by tapping the box on the palm of a hand, removing a cigarette, lighting it, inhaling, and watching the smoke as it is exhaled all contribute to the perceived need to smoke. Some go so far as to claim that they "would not know what to do with their hands" if they were to stop smoking [83]. An investigation using denicotinized cigarettes illustrated that the sensorimotor experience of smoking makes a significant contribution to the perceived satisfaction [89].
Mass media is another factor that contributes to the learning of smoking behavior. Historically, the tobacco industry recruited new smokers by associating its products with fun, excitement, sex, wealth, power, and a means of expressing rebellion and independence [90]. Such promotional efforts have proven to be especially effective on teenagers, a particularly lucrative market with a lifetime of cigarette consumption ahead of them [91]. Although at present tobacco companies can no longer directly advertise to teenagers, they retain the most potent form of marketing: movies. Smoking in film is a "more powerful force than overt advertising," perhaps because the audience is generally unaware of any sponsor involvement [92]. Philip Morris, one of the world's leading tobacco companies, stated in their 1989 marketing plan, "We believe that most of the strong, positive images for cigarettes and smoking are created by cinema and television" [90]. Although television is taking a more socially responsible stance on the subject of on-air tobacco use, movies continue to model smoking as a socially acceptable behavior, portraying it as a social behavior or a way to relieve tension [93,94]. A study exploring the connection between a child's professed favorite movie star and that actor's on-screen smoking history revealed "a clear relation between on-screen use and the initiation of smoking in the adolescents who admire them" [95]. Tobacco use in movies, albeit falling through the 1970s and 1980s, increased significantly after 1990 [90]. Furthermore, despite declining tobacco use and increasing public understanding of the dangers of nicotine, smoking in movies returned to the levels observed in the 1950s, when it was nearly twice as prevalent in society as in 2002 [96]. A study analyzing the content of the top 25 grossing films each year from 1988 to 1997 found that 87% of movies depicted tobacco use, with an average of 5 occurrences per film. The vast majority of tobacco use was portrayed as experienced use (91.5%) and rarely did it represent a character's first use (0.3%) or a relapse from a previous quit attempt (0.5%). Despite the fact that R-rated movies contained most tobacco exposure and were more likely to feature a major character using tobacco, about 60% of the total coverage of smoking occurred in youth-rated films (G, PG, and PG-13). Negative reactions to tobacco use, including comments about health effects or gestures such as coughing, were depicted in only 5.9% of the occurrences. Unrealistic portrayal of cigarette smoking on the big screen may help to explain the somewhat surprising finding that children of nonsmoking parents are especially susceptible to the effects of movie smoking exposure [93]. Between 2002 and 2017, 6 out of every 10 movies rated PG-13 contained smoking or tobacco use, with historically high average of occurrences per film in 2016 (34 per film) and 2017 (29 per film), prompting many health groups to advocate for the requirement of an R rating (i.e., younger than 17 years of age require accompanying adult) for any films containing tobacco use. Researchers estimate that requiring a R rating would reduce the number of teen smokers by 18%, preventing up to 1 million deaths from smoking in the future [184]. Since May 2007, the Motion Picture Association of America (MPAA) has made smoking a factor in assigning ratings to films. The pervasiveness of tobacco use, context in which smoking appears, and whether or not the act is glamorized are all taken into account by film raters [97].
It has been suggested that high genetic vulnerability to cigarette smoking may explain why some people begin and continue to smoke despite associated risks [98]. Twin studies found significant heritability for persistence of smoking versus quitting. Heritability estimates for smoking persistence ranged from 27% to 70% and were greater for older than younger cohorts [99,100,101]. Madden et al. examined cross-cultural differences in the genetic risk of becoming a regular smoker and of persistence in smoking in men and women. They found strong genetic influences on smoking behavior, 46% for women and 57% for men, consistent across country and age group [102]. In a U.S. study, estimates of the genetic contribution to risk of becoming a smoker were 60% in men and 51% in women [103].
Of the numerous ingredients in tobacco smoke, nicotine is believed to be the primary cause of cigarette addiction [104]. Commercially available forms of nicotine-replacement therapy (NRT) increase cessation rates approximately 1.5- to 2-fold [105,106,107]. Yet, the fact that only a fraction of those who use such products succeed suggests that cigarette addiction depends on specific characteristics of cigarette smoking. It appears that the rapid delivery of nicotine via inhalation is a primary contributor to cigarette dependence [108]. Indeed, a district court judge found that major U.S. cigarette companies have designed their cigarettes to precisely control nicotine delivery levels and provide doses of nicotine sufficient to create and sustain addiction [109].
Active components of cigarette smoke affect many organ systems, but the effects on the CNS may be of most clinical importance due to its mediating role in dependence. Central effects of nicotine include electroencephalogram (EEG) desynchronization, with a shift toward higher frequency [110]. Studies have demonstrated that nicotine from cigarette smoke reduces global cerebral blood flow (gCBF), most markedly in the right hemisphere, and increases regional cerebral blood flow (rCBF) by more than 10% in the cerebellum, occipital cortex, and insula. Decreases in rCBF have been observed in such subcortical structures as the hippocampus, anterior cingulate, amygdala, and nucleus accumbens [111]. Positron emission tomography (PET) studies show that nasal nicotine administration increases cerebral glucose metabolism in the left inferior frontal gyrus, left posterior cingulate gyrus, left lateral occipitotemporal gyrus, left and right cuneus, and right thalamus, while it decreases glucose metabolism in the left insula and the right inferior occipital gyrus [112].
Further, the physiology of nicotine dependence has been characterized as biphasic; it stimulates the pleasure response in the brain and creates a relaxed state. As with cocaine, amphetamines, and morphine, addiction to nicotine is believed to result from increased release of dopamine in the nucleus accumbens. Nicotinic acetylcholine receptors are located throughout the CNS. Neurons located in the ventral tegmental area become more active with nicotine administration, leading to an increase in dopamine release into the nucleus accumbens [113]. Indeed, lesions to these pathways reduce rates of self-administered nicotine [114].
Many smokers believe that smoking improves concentration, treats stress, and gives pleasure. These beliefs are false. The light-headed feeling that may accompany the act of smoking gives the smoker a false sense of pleasure or release. However, smoking actually causes a decline in physical and cognitive functioning. Additionally, a study by Ota et al. showed that nurses in Japan indulged in smoking as a result of the psychologic demands of their jobs, and this psychologic job demand was positively correlated with their Tobacco Dependence Screener score. The nurses associated stressful tasks with dysphoria, insomnia, anxiety, and other symptoms similar to that of nicotine withdrawal. To alleviate these symptoms, the nurses would smoke and become increasingly psychologically dependent on nicotine with each demanding occupational event [115].
Smoking severely compromises pulmonary function in a variety of ways, including causing infiltration of the airways with leukocytes. An imbalance among proteases, their endogenous inhibitors, and local cytokine secretion in the lung leads to airway inflammation and alveolar destruction. Smokers also experience more acute lower respiratory illnesses. Smoking has been implicated in the development of malignant and nonmalignant lung disease, including COPD, bronchitis, influenza, emphysema, pneumonia, and lung cancer. Smokers are also shown to be at increased risk of intraoperative pulmonary complications and a wide range of postoperative complications. For example, a study of postoperative care revealed smoking, being older than 65 years of age, and a history of chronic lung disease increased the risk of unplanned intensive care admittance [116].
Smoking is the main cause of COPD, which encompasses both chronic bronchitis and emphysema. Between 20% and 30% of smokers (or about 1 in 4) will develop COPD, and risk is determined largely based on genetic susceptibility coupled with age at smoking initiation [117,118]. It is very rare in nonsmokers; at least 80% of deaths from this disease can be attributed to cigarette smoking. The risk of death from COPD rises concurrently with the number of cigarettes smoked. If smokers with COPD quit smoking while they are still young, an improvement in lung function can be expected. However, such improvement is not possible in older people, although after cessation further deterioration will run parallel to that of nonsmokers.
The age at which one begins smoking is important. Wiencke and colleagues discovered that smoking as an adolescent causes permanent genetic changes in the lungs and forever increases the risk of lung cancer, even if the smoker subsequently stops [119]. A Canadian community health survey conducted between 2000 and 2001 found that the risks for heart disease, COPD, and rheumatoid arthritis were far higher among people who began smoking as teenagers than among their nonsmoking peers. For COPD alone, teen smokers were three times more likely to develop the condition later in life than nonsmokers. Similarly, a retrospective cohort study of adult smokers suggests that women are particularly at risk of COPD if they start to smoke before 16 years of age [120].
Upper respiratory tract infections are common, and tobacco smoke is a proven risk factor for bacterial infection. The link between influenza and smoking has been demonstrated both for adult smokers and children exposed to smoke-filled environments. According to Arcavi and Benowitz, influenza risk is higher and infections are more severe (e.g., more cough, phlegm production, breathlessness, and wheezing) in smokers versus nonsmokers. Apparently, the antibody response is depressed in cigarette smokers. Nonsmokers should also avoid SHS exposure to decrease the risk of contracting influenza [121]. In a study of Israeli military men, presence and severity of influenza was stronger in smokers than in nonsmokers. Of all smokers, 68.5% contracted influenza compared with 47.2% of nonsmokers, and 50.6% of smokers required bed rest or lost workdays compared with 30.1% of nonsmokers [122]. A 2018 study of patients older than 65 years of age showed that smokers had a higher rate of hospitalization due to influenza (47.4%) compared with nonsmokers (42.1%). In addition, the effectiveness of the influenza vaccine in preventing hospitalization was 21% among current and ex-smokers and 39% in nonsmokers [376].
Smoking is associated with a significant increase in the relative risk of pneumonia and pneumonia-related hospitalization [123,124]. Pneumonia is not only more common among smokers, it is much more likely to be fatal. Longitudinal studies have identified an increase in the mortality rate from pneumonia in smokers associated with dose-response [125]. In general, cessation of smoking is not associated with a decrease in hospitalization for pneumonia; however, patients without COPD and a greater than 10-year history of not smoking are at a decreased risk [124]. A 2013 study found that children exposed to SHS were four times more likely to develop lower respiratory illnesses, including pneumonia [126]. Proposed explanations of the increased risk for infection in active, passive, and former smokers include increased bacterial adherence, decrease of lung and nasal clearance, and changes in the immune response.
Cardiovascular disease, defined as acute myocardial infarction (MI) and stroke, is strongly related to smoking and comprises 34% of smoking-related mortality; conversely, smoking yields 16% of cardiovascular-related mortality [62]. The relative risk of MI for smokers has been estimated at 2.88 for men and 3.85 for women, and the relative risk of stroke for smokers is estimated at 2.80. These estimates do not include the effects of passive smoking. Low-tar cigarettes and smokeless tobacco have similarly been shown to increase the risk of cardiovascular events among users in comparison to nonsmokers [127]. Cigarette smoking impacts all phases of atherosclerosis, from endothelial dysfunction to acute clinical events. Both active and passive cigarette smoke exposure predispose to cardiovascular events. The exact toxic components of cigarette smoke and the mechanisms involved in smoking that are related to cardiovascular dysfunction are largely unknown, but smoking increases inflammation, thrombosis, and oxidation of low-density lipoprotein cholesterol (LDL-C). Experimental and clinical data support the hypothesis that increased oxidative exposure may be a potential mechanism for initiating cardiovascular dysfunction. Research also suggests that small doses of toxic materials from tobacco smoke cause a nonlinear dose-response effect on cardiovascular function [128]. The risk for cardiovascular disease declines rapidly after smoking is ceased [129].
Tobacco smoking is strongly related to atherosclerosis and chronic vascular disease. Atherothrombotic ischemic stroke, transient ischemic attack, and atherothrombotic origin symptomatic or asymptomatic peripheral arterial disease are all associated with a high risk of vascular death, MI, and stroke. Exposure to tobacco smoke is a noted risk factor of all these events. A positive association was found between cigarette smoking and subarachnoid hemorrhage (SAH), especially for aneurysmal SAH in women [130].
Evidence is emerging that suggests an association between the development of other neurologic diseases and smoking. A study by Riise et al. identified the risk of multiple sclerosis as higher among smokers than among those who never smoked [131].
Studies have shown that the amount of monoamine oxidase (MAO) is reduced by 30% to 40% in the brains of smokers, compared to nonsmokers or former smokers [132]. This reduction in brain MAO levels may result in an increase in levels of dopamine. It has been suggested that nicotine may have short-term protective actions against mechanisms that cause Alzheimer disease; however, the numerous toxins in cigarette smoke negate any benefit [133]. Though the risk for dementia is slightly higher in smokers, the relative risk for Alzheimer disease is unclear. A 2013 Alzheimer study using a mouse model found that smoking hastens disease onset, exacerbates amyloid pathology, and increases neuroinflammation and tau phosphorylation [133]. Further research is needed in order to better elucidate the risk.
The U.S. Preventive Services Task Force recommends annual screening for lung cancer with low-dose computed tomography in adults aged 50 to 80 years who have a 20 pack-year smoking history and currently smoke or have quit within the past 15 years. Screening should be discontinued once a person has not smoked for 15 years or develops a health problem that substantially limits life expectancy or the ability or willingness to have curative lung surgery.
(https://jamanetwork.com/journals/jama/fullarticle/2777244 Last Accessed: May 11, 2022)Level of Evidence: B (There is high certainty that the net benefit is moderate or there is moderate certainty that the net benefit is moderate to substantial.)
In the United States beginning in the early 1950s, a series of epidemiologic, biochemical, pathologic, and animal studies demonstrated a link between cigarette smoking and lung cancer. Tobacco smoking increases the risk of all histologic types of lung cancer. More than 80% to 90% of people who develop lung cancer are current or past smokers. However, not all smokers will develop lung cancer [134]. Cited reasons include the modification of lung cancer risk by previous respiratory disease. In comparison to nonsmokers, smokers are 23 times more likely to develop lung cancer if male and 13 times more likely if female. The risk of lung cancer increases directly with the number of cigarettes smoked and decreases when smoking is ceased. The most important parameter of smoking that affects lung cancer risk is the duration of smoking. Smoking low-tar cigarettes does not substantially reduce the risk of lung cancer [14].
Tobacco smoking is also causally linked to other types of cancer, including oral, oropharyngeal and nasal cavity, urinary tract, larynx, pancreas, esophageal, stomach, liver, cervix, colon, breast, endometrial, prostate, and leukemia. In most cases, the risk increases substantially with duration of smoking and amount of cigarettes/tobacco consumed. Similarly, alcohol in combination with tobacco greatly elevates the risk of many forms of cancer [14].
Smoking can lead to adverse long-term effects on bone health, rendering smokers prone to falls and fractures. Many smokers begin smoking during adolescence—a point in which bone mass is still being constructed; thus, smoking may hinder a person from reaching their maximum bone mass, leaving them fragile and prone to fractures with longer recuperation time [136]. Further, cigarette smoking has been shown to be a key risk factor for osteoporosis and unfortunately, menopausal women are at increased risk due to a loss of estrogen during this period of life. Giampietro and colleagues suggest that a genetic variation in interleukin 6 (IL6) and lipoprotein receptor-related protein 5 (LRP5) observed in smoking White women may confer risk for osteoporosis among smokers [137]. In a study of human-derived osteoblast-like cells and trabecular bone organ culture, Walker et al. demonstrated the presence of the α4 neuronal nicotinic acetylcholine receptor (nAChR) and found that nicotine modulates proliferation in a dose-dependent manner, upregulates c-fos transcription factor, and affects synthesis of osteopontin, a bone matrix protein [138].
Women who smoke prior to pregnancy are more likely to experience a delay in conception and have about 30% higher odds of infertility [139]. Further, men who smoke are at increased risk of erectile dysfunction due to decreased bioavailability of nitric oxide and damage to peripheral nerves, the vascular epithelium, and structure of corporal tissue. Smoking may also affect the quality and mobility of spermatozoa [140,141]. Ramlau-Hansen et al. report a dose-dependent relationship between smoking and sperm concentration, testosterone, luteinizing hormone (LH), and the LH/free testosterone ratio [142].
Success of assisted reproduction therapy (ART) is reduced among smoking couples. In a meta-analysis, Waylen and colleagues found that smokers undergoing ART (e.g., in-vitro fertilization, intracytoplasmic sperm injection, gamete intrafallopian transfer, zygote intrafallopian transfer) had lower odds of live birth per cycle (i.e., birth of one or more infants that show signs of life). They also observed lower odds of clinical pregnancy per cycle (i.e., a sonographically visible gestational sac in the uterus) and higher odds of spontaneous miscarriage and ectopic pregnancy when compared to nonsmokers undergoing the same treatments [143]. A retrospective study published in 2018 found that smoking has a negative effect on endometrial thickness on the day of the embryo transfer, resulting in lower rates of implantation and pregnancy [466].
If conception is achieved (with or without ART), maternal smoking during pregnancy increases the risk for adverse conditions including low birth weight, spontaneous abortion, placenta previa, abruptio placentae, preterm premature rupture of the membrane (PPROM), and overall poor outcomes [144,145].
The miscarriage rate among mothers who smoke may be as high as 33% [146,147]. This may be due to an increased syncytial necrosis and increased thickness of syncytio/cytotrophoblast membrane, as smoking appears to induce dysfunction of villous and invasive trophoblasts early in pregnancy. Additionally, maternal levels of estriol, estradiol, human chorionic gonadotropin, and human placental lactogen are lower in smokers than in nonsmokers [148]. All of these are markers of prenatal health and well-being.
There is a strong comorbidity between alcohol consumption and tobacco use. Drinkers are more likely to smoke than nondrinkers, and smokers are more likely to drink than nonsmokers [149]. In fact, smokers are 30% more likely to consume alcohol and 10 times more likely to develop alcoholism than nonsmokers. Between 80% and 95% of all persons with alcohol use disorder also smoke cigarettes, and 70% are heavy smokers who consume more than one pack per day [150]. A study examining an association between alcohol and tobacco, using a combination of short-term (1-year) and long-term (15-year) follow-up intervals, found that past-year alcohol and tobacco use disorders were associated not only cross-sectionally, but also prospectively. These associations were present even after controlling for age, gender, and race. Year 1 tobacco dependence prospectively predicted diagnosis with an alcohol use disorder (AUD) at year 2, and a baseline diagnosis of AUD increased the likelihood of diagnosis with tobacco dependence 15 years later. Having been diagnosed with tobacco dependence at year 1 predicted AUD persistence, and vice versa. These findings demonstrate the complex association between tobacco dependence and AUDs [151]. Similarly, a study examining the natural course of AUDs from adolescence to early adulthood found that daily smoking predicted future AUD when adolescent AUD and other disorders were controlled. It is possible that chronic smoking may contribute to alcohol tolerance, increasing alcohol consumption and metabolism [152].
In the instance of nonsmokers, data from a study by Romberger and Grant suggests that patterns of alcohol abuse would be similar in workers exposed to SHS; however, the severity of the alcohol abuse may be less pronounced [153].
Smoking usually precedes recreational drug use. Among those who used both cigarettes and marijuana by the 12th grade, 65% smoked cigarettes before marijuana, and 98% of those who used both cigarettes and cocaine smoked cigarettes first. Apparently, the earlier a person uses tobacco, the more likely he or she will be to experiment with cocaine, heroin, and other drugs. More than half of those who start smoking before 15 years of age use recreational drugs in their lifetime, compared to only a quarter of those who do not start smoking until 17 years of age or later. Moreover, those who start smoking before 15 years of age are seven times more likely to use cocaine than those who never smoke. Also, heavy smokers are more likely to use marijuana or harder drugs. For example, young people who smoke more than 15 cigarettes a day are twice as likely to use any recreational drug and 16 times more likely to use cocaine than those who smoke less frequently. They are also 10 times more likely to use a recreational drug and 100 times more likely to use cocaine than those who never smoked. Even heavy users of smokeless tobacco are more likely to experiment with drugs. High school students who used smokeless tobacco 20 to 30 days per month were four times more likely to concomitantly use marijuana than nonusers, and almost three times more likely to ever use cocaine [150].
Many smokers report a link between smoking and anxiety. Researchers at the National Institute on Drug Abuse hypothesized that impaired respiration and the detrimental effects of nicotine on blood vessels to the brain elucidate why those exposed to smoking are at an increased risk of developing anxiety disorders [154,467].
Smoking is shown to be highly comorbid with such psychiatric disorders as major depression, panic disorder, and schizophrenia. Cigarette smoke has other psychoactive properties apart from nicotinic receptor stimulation. For example, it inhibits MAO, which is the enzyme responsible for breaking down the biogenic amine neurotransmitters norepinephrine, serotonin, and dopamine in the brain [155,156]. Not surprisingly, the association between smoking and major depression is well established [157,158,159]. Reports of severe major depressive episodes after smoking cessation are also common, with the onset of depressive symptoms ranging from two days to six weeks after the initial abstinence from smoking [160,161]. In some cases, depression was alleviated with the use of NRT or antidepressants; in others, depressive symptoms went away after a relapse to smoking [160,162]. In a trial of smoking cessation using fluoxetine (30 mg), 7% of participants with a previous history of major depressive disorder (MDD) were diagnosed with major depressive episodes after a 10-week treatment, suggesting that a subset of smokers may be particularly at risk for developing MDD after smoking cessation [163].
In addition to relieving depressive symptoms or major depressive episodes associated with nicotine withdrawal, antidepressants may aid in long-term smoking cessation by substituting for the antidepressant effects of nicotine that help maintain smoking. They may also have a specific effect on neural pathways (e.g., MAO inhibition) or receptors (e.g., nicotinic-cholinergic receptor blockade) that underlie nicotine addiction. A 2013 Cochrane review assessed the efficacy of antidepressant medications to aid long-term smoking cessation. The majority (75) of the 90 randomized trials included in the review were of bupropion and nortriptyline. The reviewers found high-quality evidence that bupropion significantly increased long-term smoking cessation when used as the sole pharmacotherapy, and moderate-quality evidence (limited by the small number of trials and participants) that nortriptyline also significantly increased long-term cessation. The drugs' effectiveness for long-term smoking cessation was independent of their antidepressant effects, with efficacy similar to NRT [156].
Smoking could also be a risk factor for panic disorder [164,467]. A disproportionate number of persons with panic disorder smoke cigarettes compared to the general population [165]. Mild-to-moderate nicotine dependence was associated with an 11% lifetime prevalence of panic disorder, a rate approximately 2.5 times greater than in persons with no nicotine dependence. Pohl et al. found that female patients with panic disorder had significantly higher smoking prevalence at the onset of their illness than did control subjects (54% versus 35%) and that smoking prevalence for the female patients was also significantly higher than for the control subjects (40% versus 25%) [166]. Male smoking rates did not differ between patients and control subjects.
Although the cause of this comorbidity remains controversial, several explanations have been offered: smoking promotes panic by inducing respiratory abnormalities/lung disease; nicotine produces the physiologic effects characteristic of panic by releasing norepinephrine; cigarette smoking is a form of self-medication for panic disorder; and/or a shared vulnerability promotes both conditions [167]. One study examined the effect of smoking cessation on the reduction of panic symptoms by monitoring the post-cessation abstinence status of 185 smokers. Abstinence was biochemically verified at weeks 1 and 2 and month 1. The severity of panic-relevant symptoms was self-reported by the participants at month 1 and month 3, post-cessation. The 80 participants (43.2%) who remained abstinent for one month, relative to the 105 (56.8%) who did not, demonstrated significant reductions in self-reported panic symptoms [168].
Smoking is also more prevalent in persons with schizophrenia, although reasons for its pervasiveness remain debatable [169,170,171]. Investigators have suggested that nicotine might temper positive or negative symptoms, and cigarette smoking is used as self-medication (e.g., to treat cognitive impairment and anhedonia) [171,172,173,174]. Nicotine may also attenuate the adverse effects of neuroleptics, perhaps by reducing elevated blood levels after use of antipsychotic medications [128,175,176]. Weiser et al. examined the prevalence of cigarette smoking in apparently healthy adolescents later hospitalized for schizophrenia. The number of cigarettes smoked was significantly associated with the risk for schizophrenia. Compared to nonsmokers, adolescents who smoked 1 to 9 cigarettes per day were 1.38 times as likely to be hospitalized later for schizophrenia, and adolescents who smoked 10 cigarettes per day or more were 2.28 times as likely; the latter difference was statistically significant. The authors concluded that the higher prevalence of smoking in future schizophrenia patients might indicate that impaired nicotinic neurotransmission is involved in the pathophysiology of schizophrenia [177]. Bupropion has been found to increase smoking abstinence rates in smokers with schizophrenia [178]. Additionally, a number of medications that target nicotinic acetylcholine receptors have been tested or are in development, but further research is necessary to determine their clinical utility in the treatment of schizophrenia [174].
Maternal cigarette smoking before and during pregnancy adversely affects the health of both mother and fetus. However, analysis of data from the 2020 National Vital Statistics Systems (NVSS) indicated that 5.5% of pregnant women in the United States reported smoking during pregnancy; smoking during pregnancy is more common in rural and suburban America (approximately 14% and 12%, respectively) [452]. In addition to the effects on fertility and embryonic health discussed, maternal smoking before conception increases the risk of sudden infant death syndrome (SIDS), and smoking at the time of conception increases the risk of infants being born with cleft lip, with or without cleft palate [14,180]. A 2010 study showed that as many as 8% of preterm deliveries, 7% of preterm-related deaths, 19% of term low-birth-weight deliveries, and 34% of SIDS cases in the United States were attributable to prenatal smoking [181]. Further, several studies indicate that the offspring of mothers who smoked during pregnancy are at elevated risk of developing nicotine dependence as adults [182,183].
According to 2016 NVSS data, the prevalence of smoking during pregnancy was highest among women who were between 20 and 24 years of age (10.7%), followed by women 15 to 19 years of age (8.5%) and 25 to 29 years of age (8.2%). Among racial groups, the highest rates were found in non-Hispanic American Indian/Alaska Native women (16.7%%), followed by White (10.5%), Black (6.0%), Native Hawaiian/Pacific Islander (4.5%), Hispanic (1.8%), and Asian (0.6%) women. Smoking rates were highest among those with a high school diploma or equivalent (12.2%), followed by those with less than 12 years of school completed (11.7%), and women with some college or an associate's degree (7.9%). Less than 1% of women with a bachelor's degree or higher reported smoking during pregnancy [179]. Rates of maternal smoking during pregnancy differ greatly between individual states, with West Virginia (25.1%) and Kentucky (18.4%) reporting the highest percentages, and the District of Columbia (2.6%) and California (1.6%) reporting the lowest. SHS exposure in infancy greatly increases the odds of respiratory tract infections, ear infections, and death from SIDS [14].
Ohida and colleagues performed cross-sectional surveys in Japanese obstetric clinics to investigate the effects of passive smoking on sleep disturbance during pregnancy [185]. Pregnant women exposed to passive smoking were likely to have insufficient sleep, difficulty initiating sleep, short sleep duration, loud snoring, or uncomfortable breathing. These experiences also occurred in pregnant women who were smokers.
Nicotine has a low molecular weight and high lipid solubility, allowing it to cross the placenta freely and accumulate in amniotic fluid. In animal models, nicotine could be identified in fetal tissues as early as five minutes following maternal injection [186,187]. Because less than 5% of nicotine binds to human plasma proteins, the majority of the administered dose is available to equilibrate with fetal circulation [188]. Studies in humans showed that nicotine is readily transferred to the fetal compartment throughout pregnancy, with accumulation in placental tissue and amniotic fluid [189]. Apparently, a significant amount of nicotine is retained by the placenta and may later transfer to fetal and maternal circulation, thus prolonging the effect of nicotine on the fetus [188].
Acetylcholine causes dilation of blood vessels and maintains placental blood flow by the activation of endothelial muscarinic receptors. Nicotine blocks acetylcholine-facilitated amino-acid transport, depressing diffusion of amino acids and other nutrients from the trophoblast into placental circulation. Maternal smoking actually leads to trophoblast apoptosis and thickening of the trophoblast basement membrane [190,191]. Further, CO from tobacco smoke crosses the placenta by passive diffusion, leading to increased carboxyhemoglobin in umbilical cord blood and placental hypoxia. The resultant hypoxia causes fetal growth retardation and alteration in the physiologic development of organs and tissues [192].
Among pregnant smokers, maternal levels of cotinine correlate better with outcome measures such as birth weight than the number of cigarettes smoked per day [193]. Cotinine can accumulate in fetal compartments as early as 7 weeks' gestation in both active and passive smokers [194]. Of note, the half-life of nicotine is three to four times longer in newborns than in adults, whereas the half-life of cotinine is similar in newborns and adults. The prolonged elimination of nicotine, but not of cotinine, in the newborn compared with that in the adult may be a result of different newborn cytochrome P450 2A6 (CYP2A6) enzymatic substrate specificity, low CYP2A6 activity with another enzyme that is primarily responsible for cotinine metabolism, or differences in tissue distribution [195]. Also, pregnancy is well known for affecting metabolism of some drugs and may contribute to higher or lower clearances compared with the nonpregnant state [196]. Indeed, metabolic clearance of both nicotine and cotinine are substantially increased during pregnancy, resulting in a marked decrease in the half-life of cotinine. The mechanism for such increase in metabolic clearance is not known. It is possible that nicotine and cotinine clearances are accelerated by faster oxidation via CYP2A6 and faster glucuronide formation. Although nicotine and cotinine share the same metabolizing enzymes, their increased clearances may occur by different physiologic mechanisms. Nicotine is a rapidly cleared drug with a high affinity for CYP2A6, and the rate of clearance is primarily controlled by liver blood flow. Cotinine is a slowly metabolized chemical, with a low affinity for CYP2A6 relative to nicotine. The level of CYP2A6 in the liver, which is markedly elevated during pregnancy, primarily determines the rate of cotinine metabolism. A substantial increase in the percentage of nicotine and cotinine excreted as their glucuronide conjugates is also observed in pregnancy, but there is no increase in the percentage of 3'-hydroxycotinine excreted as a glucuronide. This suggests an acceleration of nicotine and cotinine metabolism via the N-glucuronidation pathway, but no effect on hydroxycotinine metabolism by the O-glucuronidation pathway. Also, the profile of nicotine and its metabolites in urine is altered during pregnancy. The excretion of nicotine is substantially decreased, and despite large differences in plasma cotinine concentration during smoking, there is no difference between the daily dose of nicotine absorbed from cigarette smoking during and after pregnancy [197].
Fetal nicotine exposure can result in permanent abnormalities of the dopaminergic regulation of the brain [198]. These effects can occur even at low nicotine doses and lead to a greater nicotine dependence [182]. Unlike in mature organisms, where stimulation of a target cell elicits only a short-term response, receptor stimulation in the developing systems interacts with the genes controlling cell differentiation, permanently altering the cells' responsiveness. Nicotine exposure to the prenatal brain may also prematurely stimulate the shift from proliferation to differentiation; thus, nicotine may act as a cholinergic signal, mimicking trophic effects of acetylcholine. Because of the close regulatory association of cholinergic and catecholaminergic systems, adverse effects of nicotine involve multiple transmitter pathways and influence not only the immediate developmental events in the fetal brain but also the eventual programming of synaptic competence. Therefore, defects may appear after a prolonged period of apparent normality, leading to cognitive and learning defects that appear in childhood or adolescence. Similar modifications occur in peripheral autonomic pathways, leading to increased susceptibility to hypoxia-induced brain damage and perinatal mortality [199]. These changes are especially prominent in tissues rich in nicotinic cholinergic receptors, such as the brainstem [200].
Prenatal exposure to nicotine produces alterations in tegmental nuclei related to the following [201]:
Cardiopulmonary integration (nucleus tractus solitarii, parabrachial complex)
Regulation of arousal, attention, and rapid eye movement (REM) sleep (mesencephalic and pontine reticular formation)
Somatic motor control (paramedian pontine and medullary reticular formation)
Tongue and upper airway regulation (hypoglossal nucleus)
Autonomic deregulation could explain the inhibition of some homeostatic reflexes seen in infants exposed to tobacco smoke, including a deficiency in arousal responsiveness to hypoxia or hypercapnia [202]. Roy et al. evaluated cellular morphology and regional architecture in the juvenile and adolescent hippocampus and the somatosensory cortex in rats prenatally exposed to nicotine. They found a substantial decrease in cell size in the hippocampal CA3 region and dentate gyrus, with corresponding decrements in cell layer thickness and increments in cell packing density. Smaller, transient changes were seen in CA1. There was a reduction in the proportion of medium-sized pyramidal neurons in layer five of the somatosensory cortex and an increase in the proportion of smaller, nonpyramidal cells. All regions showed elevated numbers of glia. These data demonstrate that prenatal nicotine exposure compromises neuronal maturation, leading to long-lasting alterations in the structure of key brain regions involved in cognition, learning, and memory [203].
Fetal growth and duration of gestation are the major factors affecting lung development [204]. Intrauterine influences that retard fetal weight gain may irrecoverably restrict the growth of the airways, with consequences persisting throughout the individual's life span. Fetal exposure to nicotine is associated with several abnormalities in lung growth. In animal studies, nicotine has been shown to directly interact with nicotinic acetylcholine receptors in pulmonary vessels, altering connective tissue expression and producing vascular structural alterations [205]. Furthermore, maternal nicotine exposure results in larger alveolar volumes and suppresses alveolarization in the lungs of the offspring of rats, reducing the surface potentially available for gas exchange [206,207]. Human smokers have a high rate of poor perfusion patterns, suggesting that smoking during pregnancy may compromise uteroplacental blood flow and contribute to poor fetal development [208,209].
Maternal smoking during pregnancy poses severe risks to the developing fetal heart. Nicotine alters cardiac cell differentiation to increase the cellular injury caused by hypoxia [210]. Prenatal nicotine exposure interferes with the ability of neonatal adrenal glands to secrete catecholamines in response to hypoxia [200]. Given that the neonatal heart lacks functional sympathetic innervation, there is virtually a complete dependence on circulating catecholamines secreted by the adrenal medulla to maintain heart rate response to hypoxia. Nicotine exposure reduces the number of cardiac ß-adrenergic receptors, magnifying functional consequences of impaired catecholamine release [211]. The resultant impaired cardiac function can lead to cardiovascular collapse, subsequent brain damage, and/or death during delivery [212,213].
Adenosine diphosphate (ADP) is a major factor in determining electrical stability of myocytes, because the longer the action potential, the higher the likelihood of abnormal cardiac activity [214]. It is possible that a component in smoke temporarily disables electrical properties of ventricular myocytes, rendering the ventricular muscle more susceptible to developing arrhythmias [215].
Fetuses exposed to smoke also manifest an increase in cardiac volume growth between 23 and 27 weeks' gestation [216,217]. This could be attributed to either an exaggeration of normal cardiac growth patterns or a compensatory response to an increase in upper body growth at the time.
Infants born to mothers who smoke weigh less than other infants (independent of maternal body mass index), and low birth weight (<2,500 grams) is a key predictor for infant mortality. Effects of maternal smoking during pregnancy on infant birth weight have been recognized since 1957; nevertheless, smoking remains the most hazardous factor affecting a newborn's weight, even at present [218,219,220]. Similar to earlier studies, Bernstein and colleagues report that maternal third-trimester cigarette smoking is one of the strongest predictors of low birth weight. This study is thought to be the first to accurately assess maternal smoking levels, and startlingly, they purport that there is an estimated 27 g reduction in birth weight per cigarette consumed each day during the third trimester, or roughly twice the amount previously shown [220]. Another study found that 11.5% of infants born to women smoking less than six cigarettes daily had low birth weight [221]. Taken together, these studies demonstrate that there is not a safe level of smoking during pregnancy [221,222]. Additionally, Aagaard-Tillery et al. reported that tobacco-exposed infants were small for gestational age regardless of maternal body mass index or pregnancies complicated by diabetes or hypertension [223].
A study examining the effect of prenatal smoke on a fetus in midgestation identified greater early gestational upper-body growth with preferential growth of head dimensions, upper limb length, and abdominal circumference with smoke exposure. This was followed by decreases in biparietal dimensions of the head, abdominal diameter, and distal limb length. Data from the late gestation period revealed cranial dolichocephaly, proportionally longer upper limbs, and legs with relatively reduced tibias, indicating that smoke exposure altered the growth rate of individual body segments [216]. It is possible that during hypoxia, blood supply to the lower limbs and internal organs is reduced in order to preserve brain metabolism [224]. Retardation of limb growth by 32 weeks could be due to diminished oxygen availability for distribution to distal tissues. The tibia, being one of the last consumers in the fetal nutrient distribution food chain, is therefore regarded as a good marker of available oxygen resources [216].
Passive smoke exposure is independently associated with an increased risk of otitis media [222,225,226]. Though the immediate complications of otitis media are significant, one must also consider the lasting complications including an increased prevalence of speech and language difficulties, attention disorders, and learning difficulty [226]. The mechanism by which cigarette smoke causes otitis media is currently unknown. Histologic changes in fetal alveolar and bronchial epithelium lend support to a contemporary theory that purports that fetal cigarette smoke exposure may interfere with the development of the middle ear and eustachian tube epithelium. An alternative theory proposes that passive smoke-related immune system depression allows for opportunistic middle ear infections [226].
One of the potentially negative effects of smoking during pregnancy is exposure of the fetus to carcinogens [227,228]. The potent tobacco-related carcinogen 4-aminobiphenyl has been shown to cross the human placenta and bind to fetal hemoglobin [229]. Two metabolites of the tobacco-specific transplacental carcinogen NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) and its glucuronide (NNAL-Gluc), were detected in the urine from newborns of mothers who smoked cigarettes during pregnancy [144]. Studies relating childhood and in utero cigarette exposure to brain tumors and leukemia have been inconsistent in their findings [230]. A meta-analysis of the association between exposure to maternal tobacco smoke during pregnancy and cancer in childhood found a small increase in risk of all neoplasms (based on 12 studies) but not of specific neoplasms such as leukemia (based on 8 studies) and CNS tumors (based on 12 studies) [231].
Maternal smoking has been shown to modulate bone mineral acquisition for the fetus, which may lead to increased risk of osteoporosis later in life [232].
Previous studies have reported an association between maternal smoking during pregnancy and behavioral problems such as hyperactivity and decreased attention span. The association with behavioral problems has been shown in investigations of hyperactive children and controls, sibling studies in which the mother smoked in one pregnancy but not in the other, and in neuropsychologic evaluations of children of smokers and nonsmokers using tests of sustained vigilance and attention [233,234,235,236]. Naeye and Peters found that hemoglobin levels in neonates increased with the number of cigarettes smoked by the mother during pregnancy and that children who were more active or had shorter attention spans had significantly higher hemoglobin levels [235]. Further, early secondhand exposure to nicotine as a child via maternal smoking during pregnancy shows an association with offspring attention deficit hyperactivity disorder (ADHD) symptoms [237,238]. Evidence also supports a statistical association between prenatal smoking and increased risk for antisocial outcomes in offspring. Maternal smoking during pregnancy has been shown to be associated with a significant increase in externalizing behavior (tendency to seek controversy, aggressive, hyperactive) but not internalizing behavior (withdrawn, depressed, anxious) problems [239]. Similarly, maternal smoking during pregnancy has been shown to have an adverse effect on the child's negativity [240]. In a sample of 99 children 2 years of age, maternal smoking was identified as a significant predictor of childhood negativity, independent of demographic factors, perinatal factors, maternal personality attributes, and the mother-child relationship. Behavior problems associated with in utero exposure to SHS seem to continue into childhood and young adolescence, demonstrated by increased risk for ADHD, conduct disorders, criminality, and substance abuse [241]. An 18-year epidemiologic study of 1,265 New Zealand children identified that maternal smoking during pregnancy contributed to risk of higher psychiatric symptom rates for conduct disorder(s), alcohol abuse, substance abuse, and depression [242,243].
The American Heart Association recommends that all children at increased risk for complications be screened for smoke exposure and provided with counseling on lowering exposure and quitting. The nicotine patch or gum can be considered if counseling is ineffective.
(https://www.ahajournals.org/doi/10.1161/CIR.0000000000000618 Last Accessed: May 11, 2022)Level of Evidence: A (Well-designed randomized controlled trials or diagnostic studies performed on a population similar to the Guidelines' target population)
It is possible that SHS exposure during childhood may be potentially more hazardous to neurodevelopment than in utero exposure to maternal smoking. Young children have higher ventilation rates, meaning they receive higher levels of SHS for the same duration and level of external exposure [244]. Passive smoking is believed to increase the prevalence of sudden infant death syndrome (SIDS); exacerbate asthma symptoms; interfere with cognition and behavior; increase cancer risk; and cause respiratory tract illness [226,245,246]. Breastfed infants with a smoking or snuff-taking mother are exposed to nicotine in breast milk, with a mean intake of nicotine of 7 mcg/kg per day [247]. Older children experience decreased physical fitness and are susceptible to tobacco-related illnesses just as adult smokers are.
Aside from adverse health effects due to SHS exposure, parental smoking is also positively correlated to their offspring's smoking as adolescents and adults. Counseling parents on the adverse health effects of SHS on children has been shown to dramatically reduce their children's subsequent cigarette smoke exposure [6,246]. Smokers should be encouraged to smoke outside their homes and minimize SHS exposure to their children [248]. However, studies have shown that, though smoking outdoors decreases SHS exposure, children of parents who smoke outdoors still have higher prevalence of ear infections and respiratory symptoms than children of nonsmokers [249].
Prenatal and perinatal exposure to SHS adversely affects neurobehavioral development. Evidence now supports the notion that tobacco-exposed infants are more excitable and hypertonic, require more handling, and show more stress and abstinence signs than infants not exposed to tobacco. Symptoms are particularly noteworthy in the CNS, gastrointestinal system, and visual areas [250]. The presumed neurobiologic effect of SHS is altered brain development resulting from fetal hypoxia, due to either nicotine acting to reduce blood flow to the fetus, or possibly CO, which produces higher levels of carboxyhemoglobin. Nicotine may also target specific neurotransmitter receptors in the fetal brain to discoordinate the events of cell replication, differentiation, and synaptic development in the brain. Nicotine is thought to disrupt brain development via cholinergic mechanisms. In rats, exposure to nicotine alone has been shown to result in a significant increase in acetylcholinesterase (AChE) activity in the brainstem and midbrain. A significant increase in ligand binding to nAChR has been observed in the brainstem and cortex following exposure to nicotine. This suggests that exposure to nicotine may impair neurobehavioral performance and affect the cholinergic pathways [251].
In another study, postnatal SHS reduced hindbrain (comprising the pons and medulla oblongata) DNA concentration, increased the protein-to-DNA ratio, and reduced the body weight of exposed rats. These data suggest that postnatal exposure to SHS affects the hindbrain, a region that undergoes significant postnatal growth, by reducing the total number of cells and by increasing cell size. The authors concluded that, despite preserved hindbrain weight, the effects of postnatal exposure to SHS might result in neurologic dysfunction [252]. This study provided clear biologic evidence for an alteration of brain development due to postnatal, but not prenatal, SHS exposure. Interestingly, although gross dysmorphology is demonstrable in the animal brain by SHS exposure to nicotine, brain structures are not grossly abnormal when examined later in adolescence or adulthood [203]. However, longer-lasting changes in morphology are noted in the hippocampus and somatosensory cortex in the form of decreased cell size and elevated numbers of glia. In considering synaptic function, several neurochemical studies have identified multiple biochemical markers of cell injury that indicate prenatal nicotine exposure damages the developing brain [253,254].
Nicotine exposure causes myocyte cell damage in newborns, reduced platelet activation, increased resting sympathetic nerve activity, and hypertension. In rats, exposure to SHS during the neonatal period resulted in abnormal vasoconstrictor and vasodilator responses and smooth muscle dysfunction [255]. Abnormalities of endothelial cell function were found in rabbits exposed to SHS for 3 to 10 weeks [256]. Exposure to SHS also appears to directly affect endothelial function in children by means of a dose-dependent decrease in the bioavailability of nitric oxide [257]. Exposure to SHS also caused left ventricular hypertrophy in rabbits [258]. SHS exposure in childhood reduces high-density lipoprotein levels [259]. In addition, adolescents exposed to their parents' smoke show depressed levels of high-density lipoprotein cholesterol (HDL-C), suggesting that SHS exposure may accelerate atherosclerotic change and place children at increased risk for the premature development of coronary artery disease [260,261].
SIDS occurs within the first year of life and is a significant cause of infant mortality, with an estimated 3,400 deaths in the United States annually [262]. SIDS is a diagnosis of exclusion, and etiology is presently unclear. Various risk factors have been suggested including prone sleeping position, sex, age, birth weight, parental cigarette smoking, maternal substance abuse, bed sharing, soft bedding, and overheating [262,263]. Matturi et al. found evidence supporting an association between maternal smoking and SIDS. Specifically, CO from cigarette smoke forms carboxyhemoglobin, leading to brain hypoxia. This lack of oxygen inhibits normal brain development of the arcuate nucleus and normal brain function in the locus coeruleus and arcuate nucleus. These abnormalities could potentially affect control of the respiratory and cardiovascular systems, resulting in sudden unexplained infant death. Matturi et al. concluded that the most preventable risk factor for SIDS is maternal smoking during pregnancy [264]. Zhang et al. concluded that the association between maternal smoking and elevated SIDS risk is dose-dependent and significantly increased in infants who co-sleep with smoking mothers [265]. Another study that sampled pericardial fluid in SIDS cases found that 70% had elevated levels of cotinine [266].
Children with smoking parents demonstrate higher frequencies of common respiratory symptoms including cough, phlegm, asthma, breathlessness, and wheeze. Parental smoking inhibits lung growth and function during childhood [267,268,269,270]. One study assessed the pulmonary function of 80 healthy infants soon after birth and found significantly reduced pulmonary function in infants whose mothers had higher urine cotinine concentrations [271]. Another study demonstrated an association between in utero nicotine exposure and variable DNA methylation in fetal lung and placental tissues, suggesting that this variation may have a role in the fetal origins of chronic diseases [272].
Both past and current SHS exposure has been shown by multiple studies to cause cough and wheeze in children. Joad et al. worked with guinea pigs to establish the mechanism by which air pollutants, particularly SHS, causes cough. Secondhand smoke modifies afferent sensory fibers (specifically C-fibers and rapidly activating receptors) in the lungs and airways, thereby activating a neurally controlled cough mechanism. The vagus nerve receives input from the afferent sensory fibers, which is modified by interneurons in the nucleus tractus solitarius (NTS). A few additional modifications of the efferent activity occur in the brain stem. Cough occurs when the efferent signal modifies input to the respiratory muscles involved in inspiration and expiration. Wheeze occurs with bronchoconstriction and mucus secretion, which can be caused by locally released neurokinins or parasympathetic fibers synapsing on airway ganglia [64].
Asthma is a chronic inflammatory disease, often with an initial onset in childhood. An association has been established between exposure to passive tobacco smoke and pediatric asthma development, while a causal relationship has been shown between exacerbated pediatric asthma and environmental tobacco exposure [273,274]. Cigarette smoke causes an "exaggerated bronchoconstrictor response" in asthmatics, leading to an increase in severity and frequency of acute asthma attacks as well as asthma-related hospitalizations [275]. Studies have shown a decreased respiratory drive and hypoxic ventilatory response in infants of smoking mothers [247]. Exposure to nicotine for the full gestation produced an increased risk of depressed hypoxic ventilatory response in rats [18]. Parents of asthmatic children should be strongly cautioned that smoke exposure is likely to dramatically worsen their child's asthma [276,277].
Each year, several billion dollars are spent treating pediatric dental caries in the United States. Dental caries are an oral infectious disease caused by Streptococcus mutans colonization and subsequent lactic acid production leading to dental decay. In addition to poverty, passive smoking is a substantial risk factor for developing dental caries. The reason for an increased prevalence of dental caries in children of low socioeconomic status is unclear. However, as poor children are more likely to be exposed to SHS, it has been suggested that environmental tobacco smoke exposure may help explain the increased dental decay in this particular population. Environmental tobacco smoke is considered a causal factor for dental caries in primary but not in permanent teeth. Mechanisms for the role of cigarette smoke in the development of pediatric dental decay include nicotine promotion of bacterial growth; immunosuppression from environmental tobacco smoke; decreased levels of vitamin C leading to increased bacterial growth; passive smoking-related saliva reduction, which impairs the natural defense against bacteria-related acid production; and a general increase in inflammation [278].
Vitamin C (ascorbic acid) deficiency is common among active smokers due to both increased metabolism and decreased dietary consumption [68]. Cigarette smoking-induced oxidant damage is caused by both the immune system's inflammatory response and free radicals in cigarette smoke. Vitamin C and other antioxidants play an important role in preventing oxidant-induced damage.
Studies have supported a dose-dependent inverse relationship between environmental tobacco smoke exposure and ascorbic acid and beta carotene concentrations [68,279]. A 2011 study found that children with no SHS exposure had higher levels of vitamin A, C, and E, beta carotene, and folate (controlling for dietary and supplement intake) than children with either moderate or high SHS exposure [279]. A lower concentration of these key nutrients was associated with higher cotinine levels. Vitamin B6, B12, and D levels were not found to be significantly affected.
The relationship between childhood passive smoke exposure and resultant health consequences in childhood has been firmly established. There is less known about the long-term respiratory effects of childhood passive smoke exposure. David et al. studied Chinese adults from the Singapore Chinese Health study who were exposed to cigarette smoke as children but never actively smoked, thereby eliminating active smoking as confounding bias often found in similar studies. They found an association, independent of adult SHS exposure, between childhood environmental tobacco smoke exposure and chronic dry cough and phlegm production. Other findings included a lack of an association between childhood SHS exposure and asthma or chronic bronchitis. Also, they found low-fiber predisposed patients to respiratory maladies [280]. One study found a 50% increase in adult-onset cancer for children whose fathers smoked, and the risk of hematopoietic cancer increased when both parents smoked [281].
Peppone et al. reported that never-smoking women who grew up with a smoking parent may have more difficulty becoming pregnant. Those exposed to SHS regularly in childhood and adulthood were 39% more likely to have suffered a miscarriage or stillbirth and 68% more likely to have trouble conceiving when trying for more than one year [282]. Further, among women exposed to environmental tobacco smoke in youth undergoing ART between 1994 and 1998, there was decrease in implantation rate and increased odds of spontaneous abortion [65].
In a study by Strohsnitter et al., early menopause was more likely to occur in never-smoking women exposed to maternal cigarette smoke. They attribute this association to smoke's effects on follicle production in utero [283].
The International Agency for Research on Cancer (IARC) Working Group concluded that secondhand tobacco smoke is carcinogenic to humans [284]. Complications of exposure to SHS include adverse effects on the pulmonary, cardiovascular, and neurologic systems as well as increased risk for cancer and fibroblast changes.
Occupational exposure to SHS affects the health of countless employees worldwide. Workplace exposure is highly influenced by the type of smoking policy in the workplace. Airborne nicotine is present, often in excessive concentrations, in various job settings due to variable public smoking laws [285,286]. Local and state regulation of smoking in public places was instituted in response to data published by the American Society of Heating, Refrigerating and Air Conditioning Engineers (ASHRAE). These standards assert that satisfactory indoor air quality cannot be maintained if smoking is allowed indoors, even with additional ventilation and air-cleaning devices [287]. Several studies have shown that smoke-free workplace policies decrease exposure of nonsmoking employees to SHS at work, while increasing rates of smoking cessation and decreasing the number of employees who smoke [14,288,289,290,291]. Policies that are less restrictive are associated with higher levels of sustained tobacco use among employees [290]. Policies that make indoor workplaces smoke-free result in improved worker health [290,292]. For example, smoke-free polices in the hospitality industry have been shown to improve health among bar workers, who are often heavily exposed to SHS in the absence of such policies [184,290,293].
Studies have shown that segregating smokers and nonsmokers within the same airspace reduces SHS exposure to nonsmokers but does not eradicate it. One such study, in smoking-segregated restaurants in Albuquerque, New Mexico, showed levels of nicotine in nonsmoking sections approximately equal to those found in smoking sections [294].
SHS remains an issue for those employed in some casinos, bowling alleys, restaurants, lounges, and bars [295]. These work environments can contain high concentrations of airborne nicotine in the air if there is a lenient smoking policy. One study found that male blue-collar workers are exposed to significantly more SHS than their counterparts in management/professional occupations [296]. Also, on average, blue-collar smokers smoke more heavily than white-collar smokers [296]. Interestingly, female blue-collar workers are far less likely to smoke than women in management/professional occupations [296]. However, women's SHS exposure is approximately equal regardless of occupation, and SHS exposure is lowest for female service industry workers.
In 1986, the National Academy of Sciences warned, "SHS (also called environmental tobacco smoke) is a hazardous substance and is the most frequent source of complaint about aircraft air quality. Because of the high concentration of SHS generated in the smoking zone, it cannot be compensated for by increased ventilation in that zone" [297]. The area, volume, and ventilation rate per smoker on an aircraft is the smallest of any workplace setting. However, essentially all airlines now prohibit smoking on their planes.
Overall, exposure to SHS in different microenvironments is based on the strength of the active source, the ventilation system, and the presence and effectiveness of air-cleaning devices. Personal SHS exposure is also affected by age, gender, and race. Constant exposure to SHS at workplaces leads to various complications to the exposed workers.
SHS is estimated to cause 5% to 30% of premature deaths from heart disease each year in the United States among nonsmokers [14,298]. A key difference between the effects of smoking on the risk of cancer compared with the risk of heart disease is that the effects on cancer develop slowly, whereas the effects of smoking on the cardiovascular system occur rapidly. Passive smoking has been shown to cause atherosclerosis in both animal and human models, increase platelet aggregation, and increase myocardial oxygen demand. Multiple epidemiologic studies have consistently found an increased relative risk of cardiac events in nonsmokers with regular SHS exposure [299,300]. Investigators demonstrated through experimentation that 30 minutes of exposure to SHS compromised the endothelial function in coronary arteries of nonsmokers so that the endothelial response of nonsmokers was identical to that of routine smokers [301].
The CDC asserts that people at risk for heart disease should avoid SHS because it can increase one's risk of acute MI. A study was conducted to verify this assertion and concluded that smoking bans at public working places correlate with a reduced morbidity from heart disease [302]. Researchers have suggested that platelet activation, endothelial dysfunction, and broad inflammation may have some relevance [303]. Another theory states that even light exposure to smoke concomitantly restricts blood vessels and allows for blood clotting. This combination raises the risk for MI.
Atherosclerosis, a chronic inflammatory atheromatous disease characterized by focal, noncircumferential, and (most often) proximal plaques, is a major underlying cause of cardiovascular disease, which continues to be the leading cause of death, accounting for 874,613 deaths in the United States in 2019 [304]. Monocytes play a key role in the pathogenesis of atherosclerosis. Monocytes migrate from the blood to the subendothelial space beneath injured endothelial cells, where they differentiate into macrophages. These subendothelial macrophages readily take up oxidized LDL, becoming "foam cells." Collections of "foam cells" are dubbed "fatty streaks" and may first appear in the aorta at 10 years of age. Fatty streaks are precursors to atherosclerotic plaques. Such plaques are advanced lesions characterized by the accumulation of lipid-rich necrotic debris and smooth muscle cells [63,305]. Triggers of endothelial cell injury include hyperlipidemia; bacterial or viral infection; oxidative stress through abnormal regulation of reactive oxygen species, hypoxia, turbulent blood flow, and shear stress; and environmental irritants, such as tobacco smoke [306].
Yuan et al. exposed transgenic human apoB-100 mice to sidestream whole smoke (SSW) (a major component of SHS) in order to study the effects of SHS on atherosclerosis. The transgenic mice received SHS exposure comparable to SHS exposure a nonsmoker would receive from a typical smoking housemate. They found a decrease in plasma HDL-C levels; a decrease in the ratio between HDL-C and triglyceride; and a decrease in ratio between HDL-C and total cholesterol. Yuan et al. noted increased lipid accretion in the aorta, heart vessels, and hepatocytes corresponding to the noted blood lipid profile alterations. Furthermore, they found increased levels of monocyte chemoattractant protein-1 (MCP-1) in blood, heart tissue, and aortic tissue. Increased numbers of macrophages were noted in arterial walls. This finding was significant as MCP-1 is a chemokine that attracts monocytes to the damaged subendothelial cells in the process of plaque formation. Decreased adiponectin monomer levels were noted in the smoke-exposed mice [63]. Adiponectin is an adipocyte-specific plasma protein with potential anti-atherogenic properties. In vitro, adiponectin suppresses the endothelial inflammatory response, the proliferation of vascular smooth muscle cells, and the transition of macrophages to foam cells [307]. Finally, based on examination of the cytokine profile, Yuan et al. determined that cigarette exposure caused a permanent pro-inflammatory state; the normal adaptive response (i.e., initial pro-inflammatory Th1 type cell-mediated response to a Th2 mediated immune response) did not occur [63].
A strong association between active smoking and coronary heart disease has been well established, and one study found a 50% to 60% increase in risk for coronary heart disease development in passive smokers [308,309]. Active and passive smoking are known to [310]:
Increase the incidence and frequency to cardiac arrhythmias
Decrease the oxygen-carrying capacity of blood
Increase the incidence of coronary artery spasm
Promote atherosclerosis, thereby increasing the risk of cardiovascular disease
Increase the incidence and tendency for thrombosis
The relationship between SHS and coronary heart disease is supported by a study that shows exposure to SHS is associated with increased inflammatory markers, including higher white blood cell counts and levels of C-reactive protein, homocysteine, fibrinogen, and oxidized LDL-C [311]. The intensity of inflammation markers was proportional to the number of years of reported exposure to SHS. Furthermore, subjects with only occasional SHS exposure also experienced increased levels of inflammatory markers, showing that even low SHS exposure is a significant concern. Increased coronary risk is mechanistically mediated by increased platelet aggregation, reduced oxygen uptake and exercise capacity, accelerated lipid peroxidation, and endothelial damage by SHS [312,313,314]. Passive smoke causes arteriosclerosis by altering cholesterol concentrations or by accelerating lipid peroxidation via reductions in serum antioxidant defense [315].
Many elements of tobacco smoke, including CO, nicotine, and polycyclic aromatic hydrocarbons, contribute to the damaging effects on the cardiovascular system. Studies of the effects of tobacco smoke on platelet sensitivity suggest that nicotine is not the sole cause of increased aggregation. Burghuber et al. compared the sensitivity of platelets to the antiaggregatory action of exogenous prostacyclin (PGI2) in nonsmokers and smokers exposed to SHS for 20 minutes. No change was observed in the smoking subjects' platelet sensitivity to PGI2 after SHS exposure, but the smokers' platelets were significantly lower than that of the nonsmoking subjects' before SHS exposure. The nonsmoking subjects experienced significant changes in sensitivity to PGI2 with reported platelet sensitivities matching those of smokers after SHS exposure [316]. However, another study by Benowitz et al. showed that smokers and abstinent smokers with nicotine patches differed significantly in platelet activity despite similar nicotine levels [317]. Thus, nicotine is not the only component of tobacco smoke that mediates increased platelet aggregation.
A British regional heart study examined 4,729 men 40 to 59 years of age and found a 50% to 60% increase in coronary heart disease caused by exposure to SHS [309]. This study is significant because most studies on the relationship between SHS and coronary heart disease either show significant risk increases or only show modest risk increases. Whincup et al. used cotinine measurements to determine passive exposure to smoking. This study noted that although high cotinine levels were associated with an excessive risk of coronary heart disease, they showed little association with the risk of stroke. Whincup et al. offered an explanation for the underestimated association between serum cotinine and coronary heart disease, in that the association tends to decrease over long follow-up periods since assessment of exposure. Finally, this study suggested that risks associated with passive smoking are widespread among nonsmokers.
The American Heart Association's Council on Cardiopulmonary and Critical Care, the Scientific Committee on Tobacco and Health in the United Kingdom, and the California Environmental Protection Agency have all concluded that SHS increases the risk of heart disease [318,319,320].
According to findings of the Health and Retirement Study, a national longitudinal study of U.S. adults 50 years of age and older and their spouses, never-smokers with spouses who were current smokers had a 42% increased risk of first stroke. Former smokers married to current smokers had a stroke risk similar to respondents who were current smokers [321].
Environmental tobacco smoke exposure is associated with respiratory symptoms, asthma, a slight impairment of lung function, and increased bronchial responsiveness [322]. A Swiss study on air pollution and lung diseases with a sample of 4,197 nonsmoking adults, showed that SHS was associated with increased risk of asthma, wheezing, bronchitis, and dyspnea [323]. Greater levels of cumulative exposure to tobacco smoke in the home and workplace are also associated with an increased risk of COPD [324]. In 2005, it was estimated that a (hypothetical) elimination of SHS in home and work environments would decrease COPD diagnoses in the United States by 18% (or 11% and 7%, respectively).
In a report by Schick and Glantz of unpublished in vivo research done by Philip Morris during the 1980s, inhaled sidestream smoke was found to be four times more toxic per gram of total particulate matter than mainstream smoke. They report that the gas/vapor phase of sidestream smoke is responsible for most of the sensory irritation and respiratory tract epithelium damage that occurs [325].
SHS is an established trigger for the onset of asthma in children, and there is growing evidence that it is also a causal factor for asthma in adult nonsmokers [326]. Finland researchers found that subjects exposed to tobacco smoke in the workplace were twice as likely to develop asthma as those who were not exposed. Health effects for adult asthmatics include asthma attacks; increased sensitivity and reduced lung function; and irritation of the eyes, nose, and throat. Exposure to cigarette smoke for just one hour can cause 20% deterioration in short-term lung function of adults with asthma [327].
Lung cancer holds the distinction as "the first disease linked definitively" to both active and passive smoking [299,328,329]. Zhong et al., based on epidemiologic studies, estimate a 30% risk of lung cancer in nonsmokers exposed to environmental tobacco smoke. Chinese women have one of the highest incidences of lung cancer in the world, yet active smoking does not appear to be a major risk factor for lung cancer in this population [328]. Smoking among Chinese women is relatively rare, and among those who do smoke, cigarette consumption is limited. However, smoking among Chinese men is especially common, so their spouses are exposed to considerable quantities of environmental tobacco smoke. Thus, nonsmoking Chinese women were an ideal population for a case-control study considering the effects of environmental tobacco smoke on lung cancer. Certain histologic types of lung cancer are more commonly associated with active smoking. The risk of developing squamous cell and small cell cancer is much higher than the risk of developing adenocarcinoma and large cell carcinoma [330]. The study by Zhong et al. showed that passive smoking also favors the development of squamous cell and small cell lung cancers over adenocarcinoma and large cell carcinoma [328].
Zhong et al. conducted a meta-analysis study on the relationship between lung cancer and environmental tobacco smoke. They found a 48% increased risk of lung cancer in nonsmoking men exposed to environmental tobacco smoke in their homes, while nonsmoking men had a 29% increased risk of lung cancer if exposed to smoke at work. A 20% increased risk of lung cancer was noted in nonsmoking women exposed to smoke in their homes, while nonsmoking women had a 15% increased risk of lung cancer if exposed to smoke at work. Furthermore, environmental tobacco smoke-exposed nonsmoking women "showed statistically significant monotonic exposure-response relationships." Finally, Zhong et al. found that childhood environmental tobacco smoke exposure did not correspond to an increased risk of lung cancer in adulthood [66].
Genetics may play an influential role in the risk of developing lung cancer from SHS exposure. Polymorphisms in the gene glutathione S-transferase (GST) M1 show a greatly increased risk of developing lung cancer with SHS exposure. GSTM1 is believed to prevent tumorigenesis by detoxifying carcinogens in tobacco smoke. Lung cancer susceptibility has been associated with anomalies in several cytochrome P450 pathways and several GST enzymes that detoxify chemical carcinogens [332,333,334,335,336]. GST enzymes are considered phase II detoxification enzymes, which conjugate glutathione to carcinogens and reactive oxygen species to detoxify them. Two of the four polymorphic gene classes of GSTs, mu (µ) and theta (θ), have been linked to tobacco-associated cancers. The GSTM1 is a variant of the mu class, which contains a null allele that may be inactivated by a deletion of DNA coding sequences [336,337]. Approximately 50% of the White populations of Europe and North America have homozygous null genotypes for the GSTM1 enzymatic activity [338]. Loss of GSTM1 enzymatic activity has been associated with increased risks of various cancers, including tobacco-associated lung cancer, head and neck cancer, larynx cancer, and bladder cancers. Bennett et al. found that SHS-exposed nonsmoking women with the null polymorphism represented 42% to 49% of the lung cancer cases [337]. Women with the homozygous null genotype have a greater risk of tobacco-associated cancer relative to men [339].
GSTTI is an isoenzyme of the theta class of GSTs, which is deactivated by a homozygous deletion in 11% to 18% of Whites [338]. United deficiency of GSTT1 and GSTM1 produces a dramatically increased risk for lung cancer in U.S. populations [340]. Kawajri et al. found that a mutant variation in exon 7 of the cytochrome P450 1A1 (CYP1A1) enzyme was associated with higher rates of lung cancer in the Japanese subjects studied [341]. CYP1A1 is known to activate carcinogenic polycyclic aromatic hydrocarbons including the benzo(a)pyrene component of tobacco smoking [342]. Rebbeck et al. found a synergistic increase in lung cancer risk with both homozygous deletions of GSTM1 and the valine allele variant of exon 7 in CYP1A1 [338].
Large-scale genome-wide association studies have identified several novel lung cancer susceptibility genes, including those on chromosomes 5p15.33, 15q24-25.1, and 6p21 [343]. The 5p15.33 region is associated with risks specific to adenocarcinoma of the lung. The 15q25 region contains three nicotine acetylcholine receptor subunit genes. Their polymorphisms have been associated with nicotine dependence [343]. Associations of the 6p21 region have not been consistently replicated among studies [343,344]. Other regions (e.g., 6q23-25, 13q31.3) have also been identified by genome-wide studies as being associated with risk of lung cancer, including some studies specific to African Americans and to those who have never smoked. Further studies are necessary to assess individual susceptibility based on the combination of polymorphisms in multiple genes [343,344,345].
Houston and colleagues questioned whether active and passive smokers are more likely than nonsmokers to develop clinically-relevant glucose intolerance or diabetes. Of 4657 participants in the Coronary Artery Risk Development In Young Adults (CARDIA) study, 16.7% developed glucose intolerance at 15-year follow-up. Incidence of glucose intolerance was highest among smokers (21.8%), followed by never-smokers with passive smoke exposure (17.2%), then previous smokers (14.4%), and was lowest for never smokers with no passive smoke exposure (11.5%). The risk among current and never smokers remained after adjustment for sociodemographic, biologic, and behavioral factors, but risk in previous smokers was similar to that in never smokers without passive smoke exposure [346]. A meta-analysis conducted by Pan et al. found that both active and passive smoking are associated with significantly increased risks of type 2 diabetes. The risk was increased in individuals who had recently quit smoking, but decreased substantially as time from quitting increased. They also identified a dose-response relation for current smoking and risk of diabetes [347].
Setty, Curhan, and Choi prospectively examined over a 14-year period (1991–2005) the relation between smoking status, duration, intensity, cessation, and exposure to SHS and incident psoriasis in 78,532 women from the Nurses' Health Study II. Prenatal and childhood exposure to passive smoke as well as current and past smoking and cumulative measures of smoking were associated with an increased risk of psoriasis. The risk of incident psoriasis among former smokers decreases nearly to that of never smokers 20 years after cessation [348].
Passive smoking is known to interfere with normal tissue repair and remodeling, though the underlying pathology is not well understood. Passive smoking has been shown to obstruct wound healing by decreasing blood flow to the damaged tissue and hindering granulation tissue formation and function. Tissue repair and remodeling is heavily reliant upon fibroblasts, which migrate to the site of damage, proliferate, and secrete cytokines, growth factors, and extracellular matrix molecules. Wong et al. found that SSW smoke causes cytoskeletal changes in fibroblasts, which may account for decreased fibroblast migration. Furthermore, excess scarring in SHS-exposed individuals is likely due to a combination of prolonged cell survival (due to a cellular stress response invoked by SHS) and the aforementioned decreased cell migration [62].
Khan and colleagues designed a case-control study to investigate a possible relation between smoking and risk of development of age-related macular degeneration (AMD) among Whites. Although many risk factors are related to AMD (e.g., aging, hypertension, family history, obesity), they found a strong association between AMD and pack years of cigarette smoking, and the odds ratio increased with the amount smoked. Smoking impairs the functioning of the retinal pigment epithelium, causing buildup on the retina and subsequent damage to Bruch's membrane. Stopping smoking was associated with reduced odds of AMD and the risk in those who had not smoked for over 20 years was comparable to nonsmokers [349].
Cervical intraepithelial neoplasm (CIN) is a precursor to cervical cancer, which is the fourth most common cause of cancer-related death in women worldwide [350]. Firmly established major risk factors for CIN include active smoking and human papillomavirus (HPV) infection. A case-control study of Taiwanese women established SHS as a major risk factor for CIN in addition to active smoking and HPV. The authors presented an indirect and a direct potential mechanism for the development of CIN following SHS exposure. CIN could be caused indirectly by immune suppression or directly by a polycyclic aromatic hydrocarbon-DNA adduct [69]. Other studies continue to suggest an association between SHS and CIN, and while these studies continue to be conducted, few have provided conclusive results [468,469].
Nonalcoholic fatty liver disease is one of the most common liver diseases in the United States. Nonalcoholic fatty liver disease covers a broad range of diseases from steatosis to nonalcoholic steatohepatitis (NASH) and can have dramatically varied underlying pathology. NASH is a significant clinical concern due to potential disease progression resulting in cirrhosis and end-stage liver disease [351]. Yuan et al. employed a mouse model transgenic for human apoB100 to consider the effect of passive smoke on cholesterol and triglyceride levels. They found no significant change in cholesterol levels with passive smoke exposure but a marked increase in triglycerides in the liver. The increased lipid accretion in hepatocytes is consistent with lipid changes seen in nonalcoholic fatty liver disease [63].
Seventy percent to 80% of nicotine is initially metabolized to cotinine, primarily by CYP2A6 [195]. Cotinine is, for the most part, metabolized to 3'-trans-hydroxycotinine, mainly by the same CYP2A6 enzyme [352]. Both nicotine and cotinine undergo N-glucuronidation; however, 3'-hydroxycotinine undergoes O-glucuronidation [353]. Cotinine is also partly metabolized to 3'-trans-hydroxycotinine by CYP2A6 [352]. Cotinine has a half-life of 15 to 20 hours, and its serum concentrations are tenfold higher than nicotine; thus, cotinine is generally used as an index of nicotine exposure [354].
Cotinine can be measured in hair, nails, blood, saliva, or urine samples. Although other biomarkers for environmental tobacco smoke exposure exist, cotinine is currently the most sensitive and specific. Such objective quantification is especially important in studies concerning passive smoke exposure in children, as parental assessment of smoke exposure is frequently unreliable [65,69,277,355,356]. SHS exposure can also be assessed through CO breath analysis, measurement of certain carcinogens (e.g., NNAL can be found in urine, blood, and nails) or benzene, or measurement of respirable suspended particulates in the air [355].
Breath analysis has improved as an assessment tool. It utilizes the monitoring of volatile organic compounds, which are predominantly bloodborne and therefore enable monitoring of different processes in the body. One study utilizing a real-time breath analyzer identified the presence of volatile organic compounds (1,3-butadiene) after SHS exposure in the breath of nonsmokers [357]. While this method of smoking analysis is improving, studies using this tool still suffer issues of sampling and lack of normalization data. Results could be skewed by participants' varying degrees of exposure to other common sources of volatile organic compounds, for example, wood smoke and automobile exhaust [358].
Studies of genetic polymorphisms of genes that modulate cell growth and proliferation provide potentially helpful biomarkers associated with long-term exposure to carcinogens and eventual tumor formation. One such biomarker used to study lung cancer in SHS-exposed patients is the tumor suppressor gene p53. The p53 gene encodes a multifactorial transcription factor that controls cellular response to DNA damage [359]. Husgafvel-Pursiainen et al. found a three- to fourfold increase in the risk of p53 mutation in SHS-exposed patients who develop lung cancer, while in long-term heavy smokers, p53 mutations are found in 50% of patients with lung tumors [360]. Furthermore, Husgafvel-Pursiainen et al. demonstrated that the majority of the p53 mutations were G:C to A:T transitions. The CpG dinucleotide sites were mutational hotspots, accounting for 50% of the reported G:C to A:T substitutions within the p53 gene. Endogenous deaminations of methylated cysteine residues or preferential carcinogen binding are proposed explanations for G:C to A:T substitutions within CpG islands. This evidence supports the role of p53 as a biomarker for both passive and active tobacco-related carcinogenesis [360].
A combination of the measurement of body fluids for cotinine and hair for nicotine, with the questionnaire and interview-derived information, seems to be the optimal method for assessing SHS exposure. Empirical studies show general concordance of reported environmental or biologic measures of SHS exposure [361]. In addition, urinary cotinine is often used for evaluation of smoking-cessation program efficacy, monitoring of pregnancy/other at-risk groups, and assessment of occupational exposure [362].
The term "thirdhand smoke," or "environmental tobacco smoke," has been and is often used synonymously with SHS, but it can be more accurately described as any airborne particulate matter originating from burning tobacco. It is comprised of both active mainstream smoke (tobacco smoke exhaled by active smokers) and sidestream smoke (smoke from the burning end of a cigarette) that is inhaled by nonsmokers, and evidence shows the possibility of harm for a significant period of time after the cigarette/tobacco product has been extinguished.
In a 2009 study by Winickoff et al., more than 80% of national survey respondents (regardless of smoking status) agreed that SHS was harmful to children, but only 43% of smokers and 65% of nonsmokers thought the same of thirdhand smoke (defined as "breathing air in a room today where people smoked yesterday") [363]. Thirdhand smoke, or any exposure to residual tobacco smoke contamination on surfaces or breathing air in a room where smoking previously occurred, can be dangerous. Unfortunately, not all smokers are cognizant of these harms. Many believe that confining smoking to one room in the home or smoking in the absence of children or even smoking outside with all household windows and doors closed is enough to protect their children. Tobacco smoke does not simply disappear after cigarettes are extinguished, and it (and other toxins) may linger even with what is perceived as adequate ventilation.
Hein and colleagues were likely the first to measure nicotine content of household dust. Nicotine has a high affinity for dust particles, and the amount of tobacco smoking that occurs in the home is highly correlated with concentration of nicotine in household dust [364]. According to a study by Matt et al., vapor components of tobacco smoke "are absorbed onto walls, furniture, clothes, toys, and other objects within 10 minutes to hours after tobacco smoke has been emitted. From there, they are re-emitted into the air over the course of hours to months" [365]. Similar to findings of a study of hair nicotine levels among children in New Zealand, whether household smokers smoked indoors in the presence of their child or attempted to limit their children's smoke exposure by smoking outside or in the children's absence, the children were not protected from exposure to nicotine in the indoor air [366]. Further, skin-to-skin contact poses additional risk as nicotine was found on the index fingers of 92% of mothers in the sample [365].
Part of the reason behind the danger of thirdhand smoke may be the lead content of tobacco smoke. According to the Environmental Protection Agency, the tobacco leaves used to make cigarettes contain radioactive lead-210. Indeed, increased blood lead levels among youth is directly associated with household smoking and house dust [367]. Mainstream smoke contains at least 58 percutaneous penetration enhancers, which are used to enhance transdermal delivery of drugs. Of these, 69% are hydrophobic or strongly hydrophobic and can therefore readily permeate the skin and likely settle in percutaneous fat for continued exposure long after the cigarette has been extinguished [368]. Further, unpublished research from Philip Morris Co. shows that 4-(methylnitrosamino)-I-(3-pyridyl)-1-butanone (NNK) forms in sidestream smoke and increases up to 200% per hour during the first six hours after cigarettes are extinguished [369]. NNK has been shown to cause an exaggerated response in microglia (causing them to attack healthy brain cells) and overall neuroinflammation, which can lead to disorders such as multiple sclerosis [370].
Oie and colleagues report that low ventilation in homes can strengthen the effects of indoor pollutants. They found that odds of bronchial obstruction among children was higher in homes where they were exposed to environmental tobacco smoke as well as dampness, textile wallpaper, and plasticizer-containing surfaces [371].
The problem is not confined to homes. In a study by Matt and colleagues, it was found that cars of people who smoked in their vehicles contained elevated levels of nicotine in dust on surfaces and in the air when compared with cars of nonsmokers [372].
Haussmann et al. performed a study of fresh versus room-aged sidestream smoke to ascertain how the different types of smoke would affect rats. Their study revealed that the room-aged smoke had decreased concentrations of smoke components such as nicotine and total particulate matter. However, levels of CO remained equal to that of the fresh smoke. The rats manifested reserve cell hyperplasia in the nose and hyperplastic and metaplastic epithelial changes in the larynx; these effects were not as profound in those exposed to the room-aged smoke [373]. Rao and colleagues found that lung tissue from mice exposed to aged and diluted sidestream smoke exhibits increased angiogenesis associated with leukocyte rolling and adhesion. This phenomenon may lead to recruitment of inflammatory cells as observed in bronchitis or asthma [374]. These research studies confirm the unpublished research of Philip Morris Co. in the early 1990s, which revealed that aged sidestream smoke is more toxic to lab animals than fresh sidestream smoke [375].
The U.S. Preventive Services Task Force recommends that clinicians ask all adults about tobacco use, advise them to stop using tobacco, and provide behavioral interventions and FDA-approved pharmacotherapy for cessation to nonpregnant adults who use tobacco.
(https://jamanetwork.com/journals/jama/fullarticle/2775287 Last Accessed: May 11, 2022)Level of Evidence: A (There is high certainty that the net benefit is substantial.)
Smoking cessation may be helpful in reducing firsthand and secondhand tobacco smoke exposure by eliminating the source: the smoker(s). Parents and caregivers of young children should receive cessation counseling and/or pharmacotherapy to quit smoking and eliminate the exposure of children to SHS. Parents should also be informed of the importance of a smoke-free environment for children and that it should be instituted before pregnancy. Pregnant women must learn that smoking will likely produce lasting adverse effects on their offspring. Furthermore, smoking parents should be aware that smoking is known to cause and exacerbate asthma, chronic serous otitis, otitis media, respiratory illness, and possibly childhood cancers. A healthcare provider is required to intervene if a child is suffering from one of these disorders. Healthcare providers are responsible for advising smoking parents about the harms of passive smoke as well as how to provide a smoke-free environment for their children [249]. There are many smoking cessation resources that may be provided to patients, including several "quitlines." These hotlines provide free telephone access to a smoking cessation counselor. The National Cancer Institute's quitline is 1-877-44U-QUIT (1-877-448-7848), and both English- and Spanish-speaking assistance is available. The National Cancer Institute also hosts a cessation live chat at https://livehelp.cancer.gov. The website https://smokefree.gov also offers support, tools, and expert advice through their app, text messaging, and social media networks. Assistance for issues unique to different subgroups, such as veterans, women, adolescents, adults older than 60 years of age, and those who speak Spanish, are also available.
Although nearly 70% of patients who smoke say they would like to quit, only 7.4% are able to do so without help [376,377]. The advice of a physician alone can increase the smoking cessation rate to 10.2% [378]. It is important for physicians to add an inquiry about smoking to the questions routinely asked while a patient's vital signs are being taken (Figure 2). Further assessment using an abbreviated form of the Fagerström Test for Nicotine Dependence can provide information about whether a patient is addicted to or physically dependent on nicotine. The Fagerström test is a question and answer test that rates an individual's nicotine dependence on a scale of 0 to 10 [379].

After the diagnosis of nicotine dependence is made, the next step is to assess the patient's readiness to change. The five-stage model for readiness to change can be applied to addictive behaviors such as smoking. The stages are precontemplation, contemplation, preparation, action, and maintenance. In the precontemplation stage, a patient does not believe that smoking is a problem or refuses to consider smoking cessation. In the contemplation stage, the patient recognizes that smoking is a problem and is thinking about quitting. During the preparation stage, the patient makes specific plans to stop smoking, such as setting a quit date and determining how smoking cessation will be accomplished. In the action stage, the patient stops smoking. Finally, the maintenance stage is marked by the patient's continued abstinence from smoking. Relapse to smoking behavior is common. Patients often cycle through the stages of change several times before reaching stable abstinence [380].
Interventions can be classified into behavioral, pharmacologic, and alternative methods. Behavioral interventions include physician advice and individual, group, and telephone- or Internet-based counseling. Pharmacologic interventions include NRT, sustained-release bupropion, and varenicline. Alternative interventions include hypnosis, acupuncture, exercise, lobeline, anxiolytics, mecamylamine, and opioid agonists [381].
According to the University of Michigan Health System, healthcare professionals should advise all tobacco users to seriously consider making a quit attempt using a clear and personalized message. Advice as brief as three minutes is effective.
(https://www.med.umich.edu/1info/FHP/practiceguides/smoking/smoking.pdf Last Accessed: May 11, 2022)Strength of Recommendation/Level of Evidence: IA (Generally should be performed based on randomized controlled trials)
Brief intervention training allows healthcare professionals to offer basic support, ensuring that all smokers who come into contact with these health professionals are able to receive help as appropriate. Brief intervention offers short-term professional input, self-help leaflets and videos, and complementary therapies. This type of information can be applicable for smokers at any level. Milch et al. compared the effects of two brief interventions against treatment as usual. The minimal intervention consisted of a smoking status vital sign stamp, which documents the patient's smoking status. The enhanced intervention consisted of a five-question form that assessed the patient's level of cessation readiness and provided cessation counseling prompts for clinicians. Medical record documentation of screening for smoking and cessation advice and self-reported patient smoking cessation rates were collected 8 to 10 months after implementation. Self-reported patient smoking cessation was higher in the enhanced intervention group (12%) compared with the minimal intervention (2%) and control (4%) groups. This demonstrated that even a short questionnaire that assessed readiness to quit and provided documentation of cessation advice improved rates of clinician cessation advice and patient smoking cessation compared with no intervention [382]. In a study by Smith and Burgess of patients admitted to the hospital with diagnoses of coronary artery disease, a minimal intervention (i.e., advice from physicians and nurses and two pamphlets) resulted in 35% of the group confirmed abstinent at 12 months [383].
The U.S. Public Health Service Clinical Practice Guideline was updated in 2018, but continues to recommend the 5 A's approach for intervening with the patient who smokes [384,470]:
Ask about smoking status
Advise to quit
Assess willingness to quit
Assist by suggesting and encouraging the use of problem-solving methods for cessation
Arrange for follow-up contacts and relapse prevention
Mullen et al. found that simple changes in question format, such as moving away from requiring "yes" or "no" answers and allowing responses such as "I used to smoke" or "I have cut down," increased smoking disclosure by 40% [385]. Every clinician should ask patients about tobacco use and advise them to quit. Abrupt smoking cessation with medical and psychologic assistance is more successful than tapering or "smoking less" [461].
Given the magnitude of tobacco use as a health risk, tobacco use status should be considered a vital sign requiring regular assessment [384,386]. Nevertheless, studies continue to find that clinicians inconsistently practice assessment of tobacco use and advice to quit smoking [387]. The third step of the Five A's approach, after asking and advising, is to assess the patient's willingness to quit. For the patient who is unwilling to quit at this time, the clinician should help increase motivation by discussing the immediate and long-term risks of continued smoking, benefits of quitting, and the patient's perceived barriers to quitting. The clinician should try to make the discussion personally relevant to the patient and include risks and benefits in addition to those related to health [384]. For the patient willing to quit, the clinician should provide assistance, such as helping the patient choose a target quit date in the near future, suggesting appropriate pharmacotherapy, providing social support, advising the patient about the nature and time course of nicotine withdrawal, recommending behavioral and cognitive coping responses to use when the patient experiences urges to smoke, and perhaps making a referral to an intensive behavioral counseling program [384]. The last of the Five A's involves arranging follow-up contact. This strategy is also based on evidence that total contact time predicts treatment outcome [384]. Follow-up contact can take the form of additional office visits, telephone calls, text messages, or even written materials sent through the mail [462]. Such contact communicates the importance of the cessation attempt, provides social support, and offers the opportunity to intercede if problems have developed. Because the risk of relapse is greatest immediately after quitting, follow-up contact ideally should begin close to the target quit date [388].
Introduced by Miller in 1983, motivational interviewing is a method of counseling designed to enhance patients' motivation to change by helping them explore and resolve their ambivalence about making the change [389]. It is a collaborative, non-confrontational, "guiding" approach. Motivational interviewing for tobacco cessation utilizes active listening to understand how the patient feels about his or her tobacco use in an effort to uncover any ambivalence [384]. The healthcare provider elicits the patient's own views regarding consequences of continuing to use tobacco and benefits of quitting and asks permission to share additional information on risks when necessary. Goals are developed collaboratively, based on the patient's current readiness to change. Originally developed as an intervention for alcohol abuse, it has shown promise as a successful strategy for smoking cessation. Lai et al. reviewed 28 studies and found that motivational interviewing yields a significant increase in quit rate, especially when conducted by primary care physicians or counselors for sessions lasting more than 20 minutes [390,391]. Further, in a randomized, controlled trial, Ruger and colleagues reported that motivational interviewing for smoking cessation actually saves money, and prevents relapse, among low-income pregnant women with $628/quality-adjusted life-year saved versus usual care [392].
Because communication with patients regarding cessation of smoking is a vital aspect of patient care, it is important that discussions and printed materials are provided in the language with which the individual is most comfortable. When there is an obvious disconnect in the communication process between the practitioner and patient due to the patient's lack of proficiency in the English language, an interpreter is required. In this multicultural landscape, interpreters are a valuable resource to help bridge the communication and cultural gap between patients and practitioners.
Interpreters are more than passive agents who translate and transmit information back and forth from party to party [393]. When they are enlisted and treated as part of the interdisciplinary clinical team, they serve as cultural brokers, who ultimately enhance the clinical encounter. When providing care for patients for whom English is a second language, the consideration of the use of an interpreter and/or patient education materials in their native language may improve patient understanding and outcomes. The American Heart Association, the American Medical Association, and the American Academy of Family Physicians produce patient education references in several languages. Primary care providers may utilize these in their interactions with patients for whom English is a second language.
Behavioral interventions are nonpharmacologic treatments delivered directly to individual smokers [388]. The main disadvantage of this approach is that relatively few smokers (about 5%) are interested in attending specific classes at any given time [394,395]. Therefore, group sessions appear to be the most cost-effective approach to delivering smoking cessation interventions [396]. Although relatively few patients want to go to classes, physicians should still have a list of referral smoking cessation clinics in their area for those smokers who express an interest in attending them and for those who have failed to respond to other approaches. Simple text, app, and web-tailored cessation messages may also be an effective alternative for behavioral support, doubling the cessation rates. This concept has been incorporated into patient support programs provided by several manufacturers of smoking cessation products [394].
There are several behavioral interventions that have empirical support, such as multicomponent coping skills training (e.g., coping response therapy, problem-focused treatment, relapse prevention training, and cognitive-behavioral therapy). This training includes social support and didactic information about nicotine dependence, withdrawal symptoms, and situations that are risks for relapse (e.g., alcohol use, negative moods, or presence of other smokers) as well as training in the use of cognitive and behavioral responses to cope with urges to smoke that reduce the risk of relapse [397,398]. Aversive therapy for smoking cessation, known as rapid smoking, involves smokers in a controlled clinical setting who deeply inhale on cigarettes at six-second intervals. Up to nine cigarettes would be smoked per treatment session to produce strong aversive reactions to cigarettes [399]. Aversive cigarette use greatly declined after the introduction of NRTs, and reviews have concluded that there is insufficient evidence to determine the efficacy of this method for smoking cessation [400,401]. Another behavioral treatment, scheduled reduced smoking, involves three weeks of gradually reduced nicotine intake. In contrast with other smoking cessation strategies involving reduction of smoking, the patient does not control when and where smoking will occur. Rather, an algorithm is used to determine when each cigarette is to be smoked based on the passage of time [402].
The first-line pharmacologic interventions for smoking cessation are NRT, bupropion, and varenicline [381,403]. However, no pharmacotherapy has been approved for use among pregnant or nursing women. The five forms of NRT available are the patch, gum, lozenge, nasal spray, and inhaler. A Cochrane review found that all commercially available forms of NRT increased the quit rate by 50% to 70%, independent of the intensity of additional support provided to the individual. Although support is beneficial, it does not appear to be essential to the success of NRT [404].
All available pharmacotherapies are safe for non-pregnant or nursing adults. In a 2016 analysis, varenicline outcomes are found to be equal to NRT plus counseling, and varenicline is also associated with a reduced risk of relapse [463]. Bupropion has the added advantage of reducing smoking cessation-related hyperphagia and weight gain. It is also an antidepressant and can ameliorate withdrawal-associated anhedonia and depression.
The nicotine transdermal system, otherwise known as the patch, releases nicotine steadily during an extended period, with blood levels rising within the first 2 to 4 hours and then remaining relatively constant between 8 and 24 hours after application, depending on the product used [405]. A number of transdermal nicotine-replacement systems are available over the counter. Prescribing information inserts for all transdermal nicotine products indicate that they should be used as part of a cessation program; yet, many patients receive the patch without any physician advice or behavioral support [406]. Adverse reactions to transdermal nicotine-replacement systems seldom cause discontinuation of therapy. Thirty percent to 50% of patients experience mild skin irritation with the patch. In most patients, rotating patch application sites can alleviate this problem. Sleep disruption is usually resolved by removing the patch at bedtime [407]. Unfortunately, use of the patch without any behavioral support is not likely to be successful.
The U.S. Food and Drug Administration adopted labeling for the patch, allowing use beyond the standard duration of eight weeks. This decision was based in part on data showing that extended-duration (24-week) transdermal nicotine therapy reduced the risk for smoking lapses and increased the likelihood of recovery to abstinence compared to the standard 8-week duration of therapy [408,409].
Nicotine chewing gum is a type of NRT that may aid in smoking cessation and/or quitting smokeless tobacco. Chewing allows nicotine to be delivered quickly into the bloodstream. Typically available in either 2- or 4-mg doses, nicotine chewing gum is expected to last one to two hours. Release of nicotine from the gum is proportional to the rate of chewing, a feature that allows for self-titration [410]. However, like the patch, nicotine gum is most successful as an adjunct to behavioral interventions. Indeed, Schneider et al. showed that merely dispensing nicotine gum resulted in a lower quit rate with active gum than with placebo treatment (8% nicotine gum, 13% placebo gum) [411].
The nicotine lozenge is similar to a hard candy. It slowly dissolves in the mouth (for 20 minutes or so) to release nicotine to the brain more quickly than the patch. Shiffman, Di Marino, and Pilliteri analyzed two trials of a 21-mg nicotine patch and 4-mg lozenge to assess the efficacy of each in heavy and dependent smokers. Both therapies were found to significantly increase six-month, continuous abstinence in heavy smokers (≥40 cigarettes per day) and the highly dependent (Fagerström score >7) [412].
A 2-mg sublingual nicotine tablet has shown efficacy in several studies and has been approved in Europe to manage nicotine withdrawal [413,414,415]. Interestingly, one study found that being married was strongly associated with smoking cessation while on this medication [416]. Sublingual tablets (2 mg) have similar pharmacokinetics to that of the 2-mg nicotine chewing gum [417]. One study of high-dependence smokers (those who smoked their first cigarette of the day within 30 minutes of waking) found that a 4-mg nicotine lozenge significantly reduced withdrawal symptoms and cravings over six weeks of treatment [418].
Nasal nicotine spray (NNS) was approved by the FDA in 1997. Available by prescription, each spray contains 0.5 mg of nicotine, and a dose is defined as one spray in each nostril. In clinical trials, subjects were allowed to take up to 5 doses/hour, with a maximum of 40 doses/day (40 mg of nicotine). The cessation rates in trials with NNS at 1 year ranged from 15% to 25% [419,420,421]. A meta-analysis of nicotine replacement suggested that NNS and the inhaler might have higher quit rates than the patch or gum [422]. Indeed, nicotine administered via nasal spray is considered to be the next fastest acting delivery method after smoking and requires 11 to 13 minutes for nicotine levels to reach peak plasma concentration [423].
The FDA also approved a nicotine inhalation system consisting of a mouthpiece and a nicotine-containing cartridge. Available with a prescription, each inhaler contains 10 mg of nicotine and 1 mg of menthol, of which 4 mg of nicotine can be extracted and 2 mg are systemically available. Shallow or deep puffing results in similar nicotine absorption. Nicotine is delivered mainly to the oral cavity, throat, and upper respiratory tract, with a minor fraction reaching the lungs. A single inhaler can be used for one 20-minute period of continuous puffing or periodic use of as many as 400 puffs per inhaler. With controlled puffing in laboratory testing, venous plasma nicotine concentrations from a single inhaler puffed 80 times for 20 minutes, averaged 8.1 mcg/L at 30 minutes. Lower concentrations of 6.4 to 6.9 mcg/L have been reported for self-administration under clinical conditions. The time to reach peak plasma concentrations varies but is always significantly longer than with cigarette delivery [424].
Quitting smoking can be a difficult process, even with use of NRT. When subjects were given denicotinized cigarettes along with IV saline or nicotine, the variable most responsible for craving satisfaction, psychologic reward, and craving reduction was the denicotinized cigarette [425]. When ad libitum smoking of preferred brands was also allowed, the combination of nicotine-less cigarette and bolus IV nicotine were the most effective in lowering craving, negative affect, and total amount smoked [89]. Sensations in the tongue, nose, back of mouth, throat, windpipe, and chest showed strong correlation between nicotine-less cigarettes and the usual brand smoked by the subjects, perhaps explaining the strong effects on smoking suppression observed [425]. Therefore, it is important to recognize that while NRT is a key part of cessation therapy, it does not address all aspects of smoking behavior. In addition, certain smoking cessation strategies, such as NRT, have been found to be less effective among women than men. Given that researchers have found that women are 31% less likely to quit smoking successfully, further studies on gender-specific smoking cessation strategies are warranted [471].
Bupropion is an atypical antidepressant that has both dopaminergic and adrenergic actions [426]. In 1998, the slow-release preparation of bupropion became available as a prescription item specifically for smoking cessation, with the trade name Zyban. This treatment could be appropriate for smokers who do not wish to use an NRT or for those whose treatment with NRT has failed. Unlike NRT, smokers begin bupropion treatment one week prior to cessation. The suggested dosage is 300 mg/day, and the duration of treatment is 7 to 12 weeks [427]. A double-blind, placebo-controlled trial randomized patients to placebo or sustained-released bupropion (50 mg twice a day, 150 mg once a day, or 150 mg twice a day) and treated them for six weeks. Smokers with active depression were excluded, though smokers with a history of depression were not. The cessation rates at the end of therapy were 10.5%, 13.7%, 18.3%, and 24.4%, respectively. Follow-up at one year suggested a continued benefit of bupropion therapy [428]. Data from a study of bupropion combined with transdermal nicotine showed high long-term quit rates with the combination therapy [429]. Discontinuation of treatment may be appropriate for individuals unable to achieve significant progress after seven weeks, as success after this point is unlikely [430].
Another effective non-nicotine therapy for smoking cessation is varenicline tartrate, a partial agonist selective for nicotine acetylcholine receptor subtypes. Released in 2006, varenicline is available in monthly dose packs (0.5 mg and 1 mg tablets) and is approved for a 12-week course of treatment [403]. Patients able to quit smoking may continue the therapy for an additional 12 weeks for increased likelihood of long-term cessation and even up to a year in certain cases, to prevent relapse; however, medication should be stopped and patients should be reassessed if the intervention has not led to smoking cessation within the initial 12 week timeframe [430,431,465]. Clinical trials reveal that varenicline may be favorable to bupropion for abstinence (44% versus 30%); the medication has also been shown to help at least 20% of patients remain smoke-free for up to one year [432,433]. Recognizing that cessation success rates increase when pharmacologic and behavioral therapies are combined, the manufacturer urges patients to combine use of varenicline with a behavioral support plan. Co-administration of varenicline and transdermal nicotine may exacerbate incidence of nausea, headache, vomiting, dizziness, dyspepsia, and fatigue. One study found varenicline alone to be more effective than other treatment options, while a meta-analysis study found that combination therapy (varenicline and NRT) was more effective than varenicline alone [434,435]. In 2021, the manufacturer of Chantix, a brand of varenicline, halted production of varenicline due to unacceptably high levels of nitrosamines [480]. In addition, all lots of 0.5-mg and 1-mg tablets of Chantix were subject to a voluntary recall. However, the FDA does not recommend that patients halt use of varenicline, and generic formulations and other brands remained available.
The two second-line drugs for smoking cessation are clonidine and nortriptyline [381]. Clonidine is an antihypertensive medication that is administered orally or transdermally. It appears to increase the smoking cessation rate by approximately 11%; however, clonidine is known to produce such side effects as dry mouth, dizziness, sedation, and orthostatic hypotension [430,436]. Clonidine has not been approved by the FDA for smoking cessation but has been used with individuals who have failed NRT or bupropion [430]. Nortriptyline is a tricyclic antidepressant that has been used to assist smoking cessation, although this is an unlabeled use [430]. A 12% improvement in cessation over controls has been reported, but the limited number of trials, combined with the adverse side effects (e.g., dry mouth, weight gain, constipation, drowsiness, sexual problems), makes nortriptyline a second-line intervention [381]. Several controlled trials have failed to show any benefit for either agent [430].
Other drugs have also been used in smoking cessation. Silver acetate, which causes cigarettes to have a bad taste, has been used as a smoking cessation aid for many years. But, there appears to be little evidence for a specific effect of silver acetate in promoting quitting [437,438]. The addition of mecamylamine, a ganglionic blocker classified as an antihypertensive agent, to transdermal nicotine replacement has been shown to improve the abstinence rate in smokers compared with use of the patch alone [439,440].
Additional pharmacotherapy options are in the development phase. A nicotine vaccine and other partial agonists for the nicotine receptors are being evaluated [441]. Interference with the liver enzymes that metabolize nicotine is another approach being tested [442].
In addition, it was found that methoxsalen, a compound used to treat skin disorders, reduces the activity of CYP2A6, the enzyme that metabolizes nicotine. This allows for more nicotine, whether from a cigarette or nicotine replacement, to be present in the blood and to remain there longer, which should minimize smokers' craving to smoke. However, methoxsalen has not been proven safe for use in humans and must undergo more trials before it can be used in a smoking cessation program [443]. Tranylcypromine (a monoamine oxidase inhibitor used to treat depression) and tryptamine (substrate of MAO) are also being investigated for this purpose [444].
The FDA has cleared a transcranial magnetic stimulation (Deep TMS) system with H4-coil for use as an aid in short-term smoking cessation in adults [482]. The outpatient procedure provides noninvasive magnetic stimulation to areas of the brain known to be associated with addiction. Approval for use as a smoking cessation therapy was based on data from a multicenter, double-blind, sham-controlled trial that evaluated the efficacy and safety of the TMS system in 262 adults. Patients included in the study had a long history of smoking (average more than 26 years) and multiple failed attempts at quitting. Patients were randomized to receive either H4 deep TMS coil or sham therapy five days per week for three weeks, followed by an additional three sessions once per week for three weeks. The primary end point was the four-week continuous quit rate at any point from the start of treatment and the follow-up visit four months thereafter.
Findings showed a continuous quit rate of 17.1% in the active TMS group, compared with 7.9% in the placebo group. Among patients with four weeks of treatment, diary records, and confirmatory urine samples, the continuous quit rate was 28.4% in the TMS group and 11.7% in the placebo group. Additionally, the number of cigarettes smoked per day (secondary end point) was statistically significantly lower in the active deep TMS arm compared with placebo. TMS has been successfully used in alcohol, tobacco, cannabis, and other substance use disorders [483,484].
Similar to all addictions, nicotine withdrawal elicits a number of clinical consequences. Desire to avoid withdrawal symptoms promotes smoking. Nicotine withdrawal may last for several weeks and include such symptoms as irritability, anxiety, depression, difficulty concentrating, weight gain, restlessness, and impatience [445]. Withdrawal effects can be elicited and observed in those exposed to secondhand smoke as well as in smokers. Intensity of these withdrawal symptoms may be related to the level of nicotine dependence. In 2020, there were an estimated 30.8 million adults that smoked cigarettes [456]. Although the prevalence of cigarette smoking continues to decline, there is some evidence that this decline is a reflection of a migration to non-cigarette products, especially e-cigarettes [446,456]
A dramatic increase in public awareness concerning the dangers of SHS has corresponded to social demand for smoking restrictions. Beginning in the 1990s, McMillen et al. found broad public support in the United States for smoking restrictions in many public places, including child care centers, hospitals, shopping malls, convenience stores, fast-food restaurants, and indoor sporting events [6]. An Irish study by Mulcahy et al. demonstrated dramatic reductions in SHS exposure following a national workplace smoking ban in Ireland. Thus, this study justified such bans given the known adverse effects of SHS, which include lung disease, heart disease, and asthma [356].
Workers suffering the detrimental effects of secondhand tobacco smoke have taken legal actions. For example, a group of 60,000 flight attendants filed a suit alleging that they had endured smoking-related illnesses from being exposed to high concentrations of environmental smoke in airplane cabins when smoking was still allowed on board [447]. Although the tobacco industry (Philip Morris, R.J. Reynolds, Brown and Williamson, the Ligett Group, and the Lorillard Group) made no admission of guilt, it established the Flight Attendant Medical Research Institute (FAMRI), a $300 million not-for-profit research institute, as a part of the settlement for flight attendants who suffered and died due to SHS exposure in air cabins. FAMRI's mission is "to sponsor scientific and medical research for the early detection and cure of diseases and medical conditions caused from exposure to tobacco smoke" [448].
Efforts to regulate tobacco products include the World Health Organization's Framework Convention on Tobacco Control (FCTC). Additionally, legislation has been passed to give the FDA regulatory authority over tobacco. The main reason for these proposals is to minimize death and disease caused by tobacco smoke by reducing the prevalence of its use and the toxicity of its products. Based on scientific studies and tobacco industry documents, it is believed that tobacco products could be made less toxic if their design, content, emissions, and manufacturing were better controlled [449].
Nationwide polls reveal broad bipartisan public support for increased taxing of tobacco [450]. State cigarette taxes have been signed into law by 53 Republican and 70 Democratic governors [451]. Since 2002, the average state cigarette tax has increased from 43.4 cents to $1.91 per pack [451,473]. In February 2009, President Obama signed a 61.66-cent federal cigarette tax increase into law, bringing the federal cigarette tax to $1.01. As of 2021, the reported average national retail price per pack of cigarettes is $8.00 [453]. Increasing the cost of tobacco not only decreases tobacco use by creating a larger economic barrier to smoking, it also motivates people to try to quit. There is a consensus that for every 10% increase in the cost per pack of cigarettes, there is a resulting 2% decrease in adult smoking, a 3.5% decrease in young adult smoking, and a 6% to 7% decrease in childhood smoking [451].
Effective behavioral and pharmacologic treatments exist and can work if they are affordable, widely available, and used properly in clinics and communities. Smoking cessation group programs have been found to be more effective than minimal treatment programs, although less intensive treatment approaches, when combined with high participation rates, can still influence larger groups. Tobacco policies have reduced cigarette consumption at work and worksite tobacco smoke exposure [454]. Innovative partnerships with public- and population-based organizations to reach smokers and reduce exposure to tobacco have been initiated. There is a high level of support for smoking restrictions in public places to protect nonsmokers from tobacco smoke [455,473]. Due to the 2009 federal tax increase, several health benefits and cost savings were projected, including an increase in the number of children alive today who will not become smokers (1.2 million) and $51.9 billion in long-term healthcare savings from fewer adult and youth smokers over the lifetimes of the adults who quit and kids who never start [451,473].
Though the state and local governments and employers provide protection from tobacco smoke at work, private homes are not subject to such regulation. Educational strategies are needed to increase awareness of personal and childhood tobacco exposure both in and out of the home. As with the business microenvironment, air quality cannot be maintained if smoking is allowed indoors, even with additional ventilation and air-cleaning devices.
The purpose of this course was to increase awareness of the various implications of tobacco use and exposure and to provide examples of healthcare assessment and treatment. It should be noted that the health complications incorporated here are only part of an exhaustive list of issues linked to tobacco smoke—more findings are uncovered each day. Changes in policy (e.g., taxation, bans in federal and other public establishments, regulation by the FDA) may spur the public to take a second look before using tobacco products or exposing themselves and friends/family to its smoke. However, it is important to continue to combat tobacco use and exposure at the primary care level at every possible opportunity. Brief intervention methods are more helpful than many realize. Further, although cigarettes have historically been implicated for the majority of health problems, it is important to be cognizant of other tobacco products' health effects and the evolving trends of tobacco use.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Text Revision. Washington, DC: American Psychiatric Association; 2022.
2. Gold MS. Drugs of Abuse: A Comprehensive Series for Clinicians: Tobacco. New York, NY: Plenum Medical Book Co.; 1995.
3. Goldsmid E (ed). The Project Gutenberg eBook of A Counter-Blaste to Tobacco, by King James I. Available at http://www.gutenberg.org/files/17008/17008-h/17008-h.htm. Last accessed May 3, 2022.
4. Kluger R. Ashes to Ashes: America's Hundred-Year Cigarette War, the Public Health, and the Unabashed Triumph of Philip Morris. New York, NY: Vintage Books; 1997.
5. Cummings KM. Programs and policies to discourage the use of tobacco products. Oncogene. 2002;21(48):7349-7364.
6. Proctor RN. The Nazi war on tobacco: ideology, evidence, and possible cancer consequences. Bull Hist Med. 1997;71(3):435-488.
7. Bachinger E, McKee M. Tobacco policies in Austria during the Third Reich. Int K Tuberc Lung Dis. 2007;11(9):1033-1037.
8. U.S. Department of Health, Education, and Welfare. Smoking and Health: Report of the Advisory Committee to the Surgeon General of the Public Health Service. 1964. Available at https://profiles.nlm.nih.gov/spotlight/nn/catalog/nlm:nlmuid-101584932X202-doc. Last accessed May 3, 2022.
9. General Services Administration. Regulations and Policies. Available at https://www.gsa.gov/about-us/regions/welcome-to-the-pacific-rim-region-9/buildings-and-facilities/california/50-united-nations-plaza-fed-office-bldg/tenants/regulations-and-policies. Last accessed May 3, 2022.
10. GovTrack.us. H.R. 1256: Family Smoking Prevention and Tobacco Control Act: 111th Congress 2009. Available at https://www.govtrack.us/congress/bills/111/hr1256. Last accessed May 3, 2022.
11. U.S. Food and Drug Administration. Deeming tobacco products to be subject to the Federal Food, Drug, and Cosmetic Act, as amended by the Family Smoking Prevention and Tobacco Control Act; restrictions on the sale and distribution of tobacco products and required warning statements for tobacco products: final rule. Fed Regist. 2016;81(90):28973-29106.
12. Centers for Disease Control and Prevention. Smoking and Tobacco Use. Fast Facts. Available at https://www.cdc.gov/tobacco/data_statistics/fact_sheets/fast_facts/. Last accessed May 3, 2022.
13. Center for Behavioral Health Statistics and Quality. 2020 National Survey on Drug Use and Health: Detailed Tables. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2021.
14. U.S. Department of Health and Human Services. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service, Office of the Surgeon General; 2014.
15. Substance Abuse and Mental Health Services Administration, Office of Applied Studies. Results from the 2011 National Survey on Drug Use and Health: Summary of National Findings. Rockville, MD: U.S. Department of Health and Human Services; 2012.
16. Campaign for Tobacco-Free Kids. Toll of Tobacco in the United States of America. Available at https://www.tobaccofreekids.org/research/factsheets/pdf/0072.pdf. Last accessed May 3, 2022.
17. Perlman SE, Chernoy C, Farley SM, et al. Exposure to secondhand smoke among nonsmokers in New York City in the context of recent tobacco control policies: current status, changes over the past decade, and national comparisons. Nicotine Tob Res. 2016;18(11):2065-2074.
18. Centers for Disease Control and Prevention. Smokeless Tobacco Use in the United States. Available at https://www.cdc.gov/tobacco/data_statistics/fact_sheets/smokeless/use_us/index.htm. Last accessed May 3, 2022.
19. Centers for Disease Control and Prevention. Secondhand Smoke. Available at https://www.cdc.gov/tobacco/basic_information/secondhand_smoke/index.htm. Last accessed May 3, 2022.
20. Ellis JA, Gwynn C, Garg RK, et al. Secondhand smoke exposure among nonsmokers nationally and in New York City. Nicotine Tob Res. 2009;11(4):362-370.
21. Centers for Disease Control and Prevention. Consumption of cigarettes and combustible tobacco—United States, 2000–2015. MMWR. 2016;65(48):1357-1363.
22. United States Government Accountability Office. Tobacco Taxes: Disparities in Rates for Similar Smoking Products Continue to Drive Market Shifts to Lower-Taxed Options. Available at https://www.gao.gov/products/GAO-14-811T. Last accessed May 3, 2022.
23. Campaign for Tobacco-Free Kids. State Cigarette Tax Rates and Rank, Date of Last Increase, Annual Pack Sales and Revenues, and Related Data. Available at https://www.tobaccofreekids.org/assets/factsheets/0099.pdf. Last accessed May 3, 2022.
24. U.S. Food and Drug Administration. Hookah Tobacco (Shisha or Waterpipe Tobacco). Available at https://www.fda.gov/tobaccoproducts/labeling/productsingredientscomponents/ucm482575.htm#references. Last accessed May 3, 2022.
25. Lipari RN, Van Horn SL. Trends in Smokeless Tobacco Use and Initiation: 2002 to 2014. Rockville, MD: Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration; 2017.
26. U.S. Department of the Treasury, Bureau of Alcohol, Tobacco and Firearms. Tobacco-Subpart B. Definitions of cigars. Federal Register. 1996;141-143.
27. Baker F, Ainsworth SR, Dye JT, et al. Health risks associated with cigar smoking. JAMA. 2000;284(6):735-740.
28. Henningfield JE, Hariharan M, Kozlowski LT. Nicotine content and health risks of cigars. JAMA. 1996;276(23):1857-1858.
29. Boffetta P, Pershagen G, Jöckel KH, et al. Cigar and pipe smoking and lung cancer risk: a multicenter study from Europe. J Natl Cancer Inst. 1999;91(8):697-701.
30. Jolly DH. Exploring the use of little cigars by students at a historically black university. Prev Chronic Dis. 2008;5(3):A82.
31. Delnevo C. Smokers' choice: what explains the steady growth of cigar use in the U.S.? Public Health Rep. 2006;121(2):116-119.
32. Campaign for Tobacco-Free Kids. The Rise of Cigars and Cigar-Smoking Harms. Available at https://www.tobaccofreekids.org/research/factsheets/pdf/0333.pdf. Last accessed May 3, 2022.
33. Kann L, Kinchen S, Shanklin SL, et al. Youth risk behavior surveillance—United States, 2017. MMWR. 2018;67(8):1-479.
34. Nasim A, Blank MD, Berry BM, Eissenberg T. Cigar use misreporting among youth: data from the 2009 Youth Tobacco Survey, Virginia. Prev Chronic Dis. 2012;9:E42.
35. Malson JL, Sims K, Murty R, Pickworth WB. Comparison of the nicotine content of tobacco used in bidis and conventional cigarettes. Tob Control. 2001;10(2):181-183.
36. Venable MJ. Bidis booming/hip cigarette cheaper, but not risk free. The Richmond Times-Dispatch. June 11, 1999.
37. Campaign for Tobacco-Free Kids. Bidis. Available at https://www.tobaccofreekids.org/assets/global/pdfs/en/IW_facts_products_bidis_overview.pdf. Last accessed May 3, 2022.
38. California State Department of Health Services. Evaluation of the health hazard of clove cigarettes. JAMA. 1988;260(24):3641-3644.
39. Malson JL, Lee EM, Murty R, Moolchan ET, Pickworth WB. Clove cigarette smoking: biochemical, physiological, and subjective effects. Pharmacol Biochem Behav. 2003;74(3):739-745.
40. Knishkowy B, Amitai Y. Water-pipe (narghile) smoking: an emerging health risk behavior. Pediatrics. 2005;116(1):e113-e119.
41. Kiter G, Uçan ES, Ceylan E, Kilinç O. Water-pipe smoking and pulmonary functions. Respir Med. 2000;94(9):891-894.
42. World Health Organization, Study Group on Tobacco Product Regulation. Waterpipe Tobacco Smoking: Health Effects, Research Needs and Recommended Actions by Regulators. Available at https://www.who.int/publications/i/item/advisory-note-waterpipe-tobacco-smoking-health-effects-research-needs-and-recommended-actions-by-regulators. Last accessed May 3, 2022.
43. Ward KD, Eissenberg T, Gray JN, Srinivas V, Wilson N, Maziak W. Characteristics of U.S. waterpipe users: a preliminary report. Nicotine Tob Res. 2007;9(12):1339-1346.
44. Sepetdjian E, Shihadeh A, Saliba NA. Measurement of 16 polycyclic aromatic hydrocarbons in narghile waterpipe tobacco smoke. Food Chem Toxicol. 2008;46(5):1582-1590.
45. Saleh R, Shihadeh A. Elevated toxicant yields with narghile waterpipes smoked using a plastic hose. Food Chem Toxicol. 2008;46(5):1461-1466.
47. Smackware. A Sketch Describing the Structure of the Hookah [Adapted]. Available at https://commons.wikimedia.org/wiki/File:Hookah-lookthrough.jpg. Last accessed May 3, 2022.
48. Hoffmann D, Hoffman I, El-Bayoumy K. The less harmful cigarette: a controversial issue: a tribute to Ernst L. Wynder. Chem Res Toxicol. 2001;14(7):767-790.
49. Haag HB, Larson PS, Finnegan JK. Effect of filtration on the chemical and irritating properties of cigarette smoke. AMA Arch Otolaryngol. 1959;69(3):261-265.
50. Brunnemann KD, Yu L, Hoffman D. Assessment of carcinogenic volatile N-nitrosamines in tobacco and in mainstream and sidestream smoke from cigarettes. Cancer Res. 1977;37(9):3218-3222.
51. Tiggelbeck D. Vapor phase modification: an underutilized technology. Proc. 3rd World Conference on Smoking and Health: Modifying the risk for the smoker. 1976 DHEW Publication No. (NIH) 76-1221:507-514.
52. Valavanidis A, Vlachogianni T, Fiotakis K. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Public Health. 2009;6(2):445-462.
53. Federal Trade Commission. "Tar," Nicotine, and Carbon Monoxide of Smoke of 1294 Varieties of Domestic Cigarettes for the Year 1998. Washington, DC: Federal Trade Commission; 2000.
54. Federal Trade Commission. Federal Trade Commission Cigarette Report for 2020. Washington, DC: Federal Trade Commission; 2021.
55. Thun MJ, Heath CW. Changes in mortality from smoking in two American Cancer Society prospective studies since 1959. Prev Med. 1997;26(4):422-426.
56. Kozlowski LT, Pillitteri JL, Sweeney CT. Misuse of "light" cigarettes by means of vent blocking. J Subst Abuse. 1994;6(3):333-336.
57. Kozlowski LT, Pope MA, Lux JE. Prevalence of the misuse of ultra-low-tar cigarettes by blocking filter vents. Am J Public Health. 1988;78(6):694-695.
58. Kozlowski LT, Frecker RC, Khouw V, Pope MA. The misuse of "less-hazardous" cigarettes and its detection: hole blocking of ventilated filters. Am J Public Health. 1980;70(11):1202-1203.
59. Byrd GD, Robinson JH, Caldwell WS, deBethizy JD. Comparison of measured and FTC-predicted nicotine uptake in smokers. Psychopharmacology (Berl). 1995;122(2):95-103.
60. Adams JD, Lee SJ, Hoffmann D. Carcinogenic agents in cigarette-smoke and the influence of nitrate in their formation. Carcinogenesis. 1984;5(2):221-223.
61. U.S. Department of Health and Human Services, Office on Smoking and Health. The Health Consequences of Smoking: The Changing Cigarette. A Report of the Surgeon General. Rockville, MD: U.S. Public Health Service, Office of the Surgeon General; 1981.
62. Wong LS, Green HM, Feugate JE, Yadav M, Nothnangel EA, Martins-Green M. Effects of "second-hand" smoke on structure and function of fibroblasts, cells that are critical for tissue repair and remodeling. BMC Cell Biol. 2004;5(1):13.
63. Yuan H, Wong LS, Bhattacharya M. The effects of second-hand smoke on biological processes important in atherogenesis. BMC Cardiovasc Disord. 2007;7:1.
64. Joad JP, Sekizawa S, Chen CY, Bonham AC. Air pollutants and cough. Pulm Pharmacol Ther. 2007;20(4):347-354.
65. Meeker JD, Missmer SA, Cramer DW, Hauser R. Maternal exposure to second-hand tobacco smoke and pregnancy outcome among couples undergoing assisted reproduction. Hum Reprod. 2007;22(2):337-345.
66. Zhong L, Goldberg MS, Parent ME, Hanely JA. Exposure to environmental tobacco smoke and the risk of lung cancer: a meta-analysis. Lung Cancer. 2000;27(1):3-18.
67. Invernizzi G, Ruprecht A, Mazza R, et al. Particulate matter from tobacco versus diesel car exhaust: an educational perspective. Tobacco Control. 2004;13(3):219-221.
68. Preston AM, Rodriguez C, Rivera CE, Sahai H. Influence of environmental tobacco smoke on vitamin C status in children. Am J Clin Nutr. 2003;77(1):167-172.
69. Tsai HT, Tsai YM, Yang SF, et al. Lifetime cigarette smoke and second-hand smoke and cervical intraepithelial neoplasm— a community-based case-control study. Gynecol Oncol. 2007;105(1):181-188.
70. Lans C, Harper T, Georges K, Bridgewater E. Medicinal and ethnoveterinary remedies of hunters in Trinidad. BMC Complement Altern Med. 2001;1:10.
71. Sandler RS, Sandler DP, McDonnell CW, Wurzelmann JI. Childhood exposure to environmental tobacco smoke and the risk of ulcerative colitis. Am J Epidemiol. 1992;135(6):603-8.
72. McCormick AA, Kumagai MH, Hanley K, et al. Rapid production of specific vaccines for lymphoma by expression of the tumor-derived single-chain Fv epitopes in tobacco plants. Proc Natl Acad Sci USA. 1999;96(2):703-708.
73. Ma S, Huang Y, Yin Z, Menassa R, Brandle JE, Jevnikar AM. Induction of oral tolerance to prevent diabetes with transgenic plants requires glutamic acid decarboxylase (GAD) and IL-4. Proc Natl Acad Sci USA. 2004;101(15):5680-5685.
74. Webster DE, Cooney ML, Huang Z, et al. Successful boosting of a DNA measles immunization with an oral plant-derived measles virus vaccine. J Virol. 2002;76(15):7910-7912.
76. Pearson MG, Chamberlain MJ, Morgan WK, Vinitski S. Regional deposition of particles in the lung during cigarette smoking in humans. J Appl Physiol. 1985;59(6):1828-1833.
77. Adams L, Lee C, Rawbone R, Guz A. Patterns of smoking: measurement and variability in asymptomatic smokers. Clin Sci (Lond). 1983;65(4):383-392.
78. Darby TD, McNamee JE, van Rossum JM. Cigarette smoking pharmacokinetics and its relationship to smoking behaviour. Clin Pharmacokinet. 1984;9(5):435-449.
79. Grant SG, Woodman G, Newman SP, Pavia D, Clarke SW. Sensory mechanisms in the upper respiratory tract affect the inhalation of cigarette smoke in man. Clin Sci (Lond). 1986;71(1):117-119.
80. Ferron GA. The size of soluble aerosol particles as a function of the humidity of the air: application to the human respiratory tract.J Aerosol Sci. 1977;8(4):251-267.
81. Centers for Disease Control and Prevention. NIOSH Spirometry Training Guide. Available at https://www.cdc.gov/niosh/docs/2004-154c/pdfs/2004-154c.pdf. Last accessed May 3, 2022.
82. Lunell E, Bergström M, Antoni G, Långström B, Nordberg A. Nicotine deposition and body distribution from a nicotine inhaler and a cigarette studied with positron emission tomography. Clin Pharmacol Ther. 1996;59(5):593-594.
84. Le Houezec J. Role of nicotine pharmacokinetics in nicotine addiction and nicotine replacement therapy: a review. Int J Tuberc Lung Dis. 2003;7(9):811-819.
85. Bandura A. Influence of models' reinforcement contingencies on the acquisition of imitative responses. J Pers Soc Psychol. 1965;1(6):589-595.
86. Turner L, Mermelstein R, Flay B. Individual and contextual influences on adolescent smoking. Ann NY Acad Sci. 2004;1021:175-197.
87. Carmelli D, Swan GE, Robinette D, Fabsitz R. Genetic influence on smoking: a study of male twins. N Engl J Med. 1992;327(12):829-833.
88. Swan GE. Implications of genetic epidemiology for the prevention of tobacco use. Nicotine Tob Res. 1999;1(Suppl 1):S49-S56.
89. Rose JE, Behm FM, Westman EC, Bates JE, Salley A. Pharmacologic and sensorimotor components of satiation in cigarette smoking. Pharmacol Biochem Behav. 2003;76(2):243-250.
90. Mekemson C, Glantz SA. How the tobacco industry built its relationship with Hollywood. Tob Control. 2002;11(Suppl 1):181-191.
91. Pierce JP, Choi WS, Gilpin EA, Farkas AJ, Berry CC. Tobacco industry promotion of cigarettes and adolescent smoking. JAMA. 1998;279(7):511-515.
92. Glantz SA. Smoking in movies: a major problem and a real solution. Lancet. 2003;362(9380):258-259.
93. Dalton MA, Tickle JJ, Sargent JD, Beach ML, Ahrens MB, Heatherton TF. The incidence and context of tobacco use in popular movies from 1988 to 1997. Prev Med. 2002;34(5):516-523.
94. Dalton MA, Sargent JD, Beach ML, et al. Effect of viewing smoking in movies on adolescent smoking initiation: a cohort study. Lancet. 2003;362(9380):281-285.
95. Tickle JJ, Sargent JD, Dalton MA, Beach ML, Heatherton TF. Favourite movie stars, their tobacco use in contemporary movies, and its association with adolescent smoking. Tob Control. 2001;10(1):16-22.
96. Glantz SA, Kacirk KW, McCulloch C. Back to the future: smoking in movies in 2002 compared with 1950 levels. Am J Public Health. 2004;94(2):261-263.
97. Associated Press. MPAA Makes Smoking Bigger Factor in Ratings. Available at https://www.today.com/id/18601051#.UXF_GEoVTyM. Last accessed May 3, 2022.
98. Pomerleau CS. Co-factors for smoking and evolutionary psychobiology. Addiction. 1997;92(4):397-408.
99. Heath AC, Martin NG. Genetic models for the natural history of smoking: evidence for a genetic influence on smoking persistence. Addict Behav. 1993;18(1):19-34.
100. Madden PA, Heath AC, Pedersen NL, Kaprio J, Koskenvuo MJ, Martin NG. The genetics of smoking persistence in men and women: a multicultural study. Behav Genet. 1999;29(6):423-431.
101. True WR, Heath AC, Scherrer JF, et al. Genetic and environmental contributions to smoking. Addiction. 1997;92(10):1227-1287.
102. Madden PA, Pedersen NL, Kaprio J, Koskenvuo MJ, Martin NG. The epidemiology and genetics of smoking initiation and persistence: crosscultural comparisons of twin study results. Twin Res. 2004;7(1):82-97.
103. Heath AC, Cates R, Martin NG, et al. Genetic contribution to risk of smoking initiation: comparisons across birth cohorts and across cultures. J Subst Abuse. 1993;5(3):221-246.
104. U.S. Department of Health and Human Services. The Health Consequences of Smoking: Nicotine Addiction: A Report of the Surgeon General. Rockville, MD: Office on Smoking and Health; 1988.
105. Silagy C, Lancaster T, Stead L, Mant D, Fowler G. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2002;(4):CD000146.
106. Stead LF, Perera R, Bullen C, Mant D, Lancaster T. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2008;(1):CD000146.
107. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2014;11:CD000146.
108. Russell MA, Feyerabend C. Cigarette smoking: a dependence on high-nicotine boli. Drug Metab Rev. 1978;8(1):29-57.
109. United States of America v. Philip Morris USA Inc., f/k/a Philip Morris Inc., et al. Civil Action No. 99-2496 (GK), Filed September 01, 2005.
110. Edwards JA, Warburton DM. Smoking, nicotine and electrocortical activity. Pharmacol Ther. 1982;19(2):147-164.
111. Domino EF, Ni L, Xu Y, Koeppe RA, Guthrie S, Zubieta JK. Regional cerebral blood flow and plasma nicotine after smoking tobacco cigarettes. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(2):319-327.
112. Domino EF, Minoshima S, Guthrie SK, et al. Effects of nicotine on regional cerebral glucose metabolism in awake resting tobacco smokers. Neuroscience. 2000;101(2):277-282.
113. Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390(6658):401-404.
114. Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653(1-2):278-284.
115. Ota A, Yasuda N, Okamoto Y, et al. Relationship of job stress with nicotine dependence of smokers—a cross-sectional study of female nurses in a general hospital. J Occup Health. 2004;46(3):220-224.
116. Moller AM, Maaløe R, Pedersen T. Postoperative intensive care admittance: the role of tobacco smoking. Acta Anaesthesiol Scand. 2001;45(3):345-348.
117. Mayo Clinic. COPD. Available at https://www.mayoclinic.org/diseases-conditions/copd/symptoms-causes/syc-20353679. Last accessed May 3, 2022.
119. Wiencke JK, Thurston SW, Kelsey KT, et al. Early age at smoking initiation and tobacco carcinogen DNA damage in the lung.J Natl Cancer Institute. 1999;91(7):614-619.
120. Patel BD, Luben RN, Welch AA, et al. Childhood smoking is an independent risk factor for obstructive airways disease in women. Thorax. 2004;59(8):682-686.
121. Arcavi L, Benowitz NL. Cigarette smoking and infection. Arch Intern Med. 2004;164(20):2206-2216.
122. Kark JD, Lebiush M, Rannon L. Cigarette smoking as a risk factor for epidemic a(H1N1) influenza in young men. N Engl J Med. 1982;307(17):1042-1046.
123. Almirall J, González CA, Balanzó X, Bolíbar I. Proportion of community-acquired pneumonia cases attributable to tobacco smoking. Chest. 1999;116(2):375-379.
124. Cecere LM, Williams EC, Sun H, et al. Smoking cessation and the risk of hospitalization for pneumonia. Respir Med. 2012;106(7):1055-1062.
125. Lange P, Vestbo J, Nyboe J. Risk factors for death and hospitalization from pneumonia: a prospective study of a general population. Eur Respir J. 1995;8(10):1694-1698.
126. Charoenca N, Kungskulniti N, Tipayamongkholgul M, et al. Determining the burden of secondhand smoke exposure on the respiratory health of Thai children. Tob Induc Dis. 2013;11(1):7.
127. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43(10):1731-1737.
128. Menza MA, Grossman N, Van Horn M, Cody R, Forman N. Smoking and movement disorders in psychiatric patients. Biol Psychiatry. 1991;30(2):109-115.
129. Ong MK, Glantz SA. Cardiovascular health and economic effects of smoke free workplaces. Am J Med. 2004;117(1):32-38.
130. Can A, Castro VM, Ozdemir YH, et al. Association of intracranial aneurysm rupture with smoking duration, intensity, and cessation. Neurology. 2017;89(13):1408-1415.
131. Riise T, Nortvedt MW, Ascherio A. Smoking is a risk factor for multiple sclerosis. Neurology. 2003;61(8):1122-1124.
132. Fowler JS, Volkow ND, Wang GJ, et al. Inhibition of monoamine oxidase B in the brains of smokers. Nature. 1996;379(6567):733-736.
133. Moreno-Gonzalez I, Estrada LD, Sanchez-Mejias E, Soto C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer's disease. Nat Commun. 2013;4:1495.
134. Centers for Disease Control and Prevention. Lung Cancer: What Are the Risk Factors for Lung Cancer? Available at https://www.cdc.gov/cancer/lung/basic_info/risk_factors.htm. Last accessed May 3, 2022.
135. Centers for Disease Control and Prevention. Tobacco Use by Youth is Rising: E-Cigarettes Are the Main Reason. Available at https://www.cdc.gov/vitalsigns/youth-tobacco-use. Last accessed May 3, 2022.
136. Davis JL. The Effects of Smoking on Bone Health: Tips to Help You Quit Smoking. Available at https://www.webmd.com/osteoporosis/features/smoking-cigarettes. Last accessed May 3, 2022.
137. Giampietro PF, McCarty C, Mukesh B, et al. The role of cigarette smoking and statins in the development of postmenopausal osteoporosis: a pilot study utilizing the Marshfield Clinic Personalized Medicine Cohort. Osteoporos Int. 2010;21(3):467-477.
138. Walker LM, Preston MR, Magnay JL, Thomas PB, El Haj AJ. Nicotinic regulation of c-fos and osteopontin expression in human-derived osteoblast-like cells and human trabecular bone organ culture. Bone. 2001;28(6):603-608.
139. Centers for Disease Control and Prevention. Smoking During Pregnancy. Available at https://www.cdc.gov/tobacco/basic_information/health_effects/pregnancy/index.htm. Last accessed May 3, 2022.
140. Tostes RC, Carneiro FS, Lee AJ, et al. Cigarette smoking and erectile dysfunction: focus on NO bioavailability and ROS generation. J Sex Med. 2008;5(6):1284-1295.
141. Pasqualotto FF, Umezu FM, Salvador M, Borges E Jr, Sobreiro BP, Pasqualotto EB. Effect of cigarette smoking on antioxidant levels and presence of leukocytospermia in infertile men: a prospective study. Fertil Steril. 2008;90(2):278-283.
142. Ramlau-Hansen CH, Thulstrup AM, Aggerholm AS, Jensen MS, Toft G, Bonde JP. Is smoking a risk factor for decreased semen quality? A cross-sectional analysis. Hum Reprod. 2007;22(1):188-196.
143. Waylen AL, Metwally M, Jones GL, Wilkinson AJ, Ledger WL. Effects of cigarette smoking upon clinical outcomes of assisted reproduction: a meta-analysis. Hum Reprod Update. 2009;15(1):31-44.
144. Lackmann GM, Salzberger U, Töllner U, Chen M, Carmella SG, Hecht SS. Metabolites of a tobacco-specific carcinogen in urine from newborns. J Natl Cancer Inst. 1999;91(5):459-465.
145. Castles A, Adams EK, Melvin CL, Kelsch C, Boulton ML. Effects of smoking during pregnancy: five meta-analyses. Am J Prev Med. 1999;16(3):208-215.
146. Naeye RL. Abruptio placentae and placenta previa: frequency, perinatal mortality, and cigarette smoking. Obstet Gynecol. 1980;55:701-704.
147. Ness RB, Grisso JA, Hirschinger N, et al. Cocaine and tobacco use and the risk of spontaneous abortion. N Engl J Med. 1999;340(5):333-339.
148. Shiverick KT, Salafia C. Cigarette smoking and pregnancy I: ovarian, uterine, and placental effects. Placenta. 1999;20(4):265-272.
149. Bien TH, Burge R. Smoking and drinking: a review of the literature. Int J Addict. 1990;25(12):1429-1454.
150. Campaign for Tobacco-Free Kids. Smoking and Other Drug Use. Available at https://tobaccofreekids.org/research/factsheets/pdf/0106.pdf. Last accessed May 3, 2022.
151. Jackson KM, Sher KJ, Wood PK, Bucholz KK. Alcohol and tobacco use disorders in a general population: short-term and long-term associations from the St. Louis epidemiological catchment area study. Drug Alcohol Depend. 2003;71(3):239-253.
152. Rohde P, Lewinsohn PM, Kahler CW, Seeley JR, Brown RA. Natural course of alcohol use disorders from adolescence to young adulthood. J Am Acad Child Adolesc Psychiatry. 2001;40(1):83-90.
153. Romberger DJ, Grant K. Alcohol consumption and smoking status: the role of smoking cessation. Biomed Pharmacother. 2004;58(2):77-83.
154. Johnson JG, Cohen P, Pine DS, Klein DF, Kasen S, Brook JS. Association between cigarette smoking and anxiety disorders during adolescence and early adulthood. JAMA. 2000;284(18):2348-2351.
155. Quattrocki E, Baird A, Yurgelun-Todd D. Biological aspects of the link between smoking and depression. Harv Rev Psychiatry. 2000;8(3):99-110.
156. Hughes JR, Stead LF, Hartmann-Boyce J, Cahill K, Lancaster T. Antidepressants for smoking cessation. Cochrane Database Syst Rev. 2014;1:CD000031.
157. Carmody TP. Affect regulation, nicotine addiction, and smoking cessation. J Psychoactive Drugs. 1989;21(3):331-342.
158. Covey LS, Glassman AH, Stetner F. Cigarette smoking and major depression. J Addict Dis. 1998;17(1):35-46.
159. Glassman AH. Cigarette smoking: implications for psychiatric illness. Am J Psychiatry. 1993;150(4):546-553.
160. Stage KB, Glassman AH, Covey LS. Depression after smoking cessation: case reports. J Clin Psychiatry. 1996;57(10):467-469.
161. Vázquez FL, Becoña E. Treatment of major depression associated with smoking cessation. Acta Psychiatr Scand. 1998;98(6):507-508.
162. Bock BC, Goldstein MG, Marcus BH. Depression following smoking cessation in women. J Subst Abuse. 1996;8(1):137-144.
163. Borrelli B, Niaura R, Keuthen NJ, et al. Development of major depressive disorder during smoking-cessation treatment. J Clin Psychiatry. 1996;57(11):534-538.
164. Amering M, Bankier B, Berger P, Griengl H, Windhaber J, Katschnig H. Panic disorder and cigarette smoking behavior. Compr Psychiatry. 1999;40(1):35-38.
165. Breslau N, Kilbey M, Andreski P. Nicotine dependence, major depression, and anxiety in young adults. Arch Gen Psychiatry. 1991;48(12):1069-1074.
166. Pohl R, Yeragani VK, Balon R, Lycaki H, McBride R. Smoking in patients with panic disorder. Psychiatry Res. 1992;43(3):253-262.
167. Cosci F, Knuts IJ, Abrams K, Griez EJ, Schruers KR. Cigarette smoking and panic: a critical review of the literature. J Clin Psychiatry. 2010;71(5):606-615.
168. Farris SG, Allan NP, Morales PC, Schmidt NB, Zvolensky MJ. Does successful smoking cessation reduce anxious arousal among treatment-seeking smokers? J Anxiety Disord. 2015;36:92-98.
169. Diwan A, Castine M, Pomerleau CS, Meador-Woodruff JH, Dalack GW. Differential prevalence of cigarette smoking in patients with schizophrenic vs. mood disorders. Schizophr Res. 1998;33(1-2):113-118.
171. Ziedonis DM, Kosten TR, Glazer WM, Frances RJ. Nicotine dependence and schizophrenia. Hosp Community Psychiatry. 1994;45(3):204-206.
172. Hall RG, Duhamel M, McClanahan R, et al. Level of functioning, severity of illness, and smoking status among chronic psychiatric patients. J Nerv Ment Dis. 1995;183(7):468-471.
173. Brown RW, Maple AM, Perna MK, Sheppard AB, Cope ZA, Kostrzewa RM. Schizophrenia and substance abuse comorbidity: nicotine addiction and the neonatal quinpirole model. Dev Neurosci. 2012;34(2-3):140-151.
174. Boggs DL, Carlson J, Cortes-Briones J, Krystal JH, D'Souza DC. Going up in smoke? A review of nAChRs-based treatment strategies for improving cognition in schizophrenia. Curr Pharm Des. 2014;20(31):5077-5092.
175. Decina P, Caracci G, Sandik R, Berman W, Mukherjee S, Scapicchio P. Cigarette smoking and neuroleptic-induced parkinsonism. Biol Psychiatry. 1990;28(7):502-508.
176. Sandyk R. Cigarette smoking: effects on cognitive functions and drug-induced parkinsonism in chronic schizophrenia. Int J Neurosci. 1993;70(3-4):193-197.
177. Weiser M, Reichenberg A, Grotto I, et al. Higher rates of cigarette smoking in male adolescents before the onset of schizophrenia: a historical-prospective cohort study. Am J Psychiatry. 2004;161(7):1219-1223.
178. Tsoi DT, Porwal M, Webster AC. Interventions for smoking cessation and reduction in individuals with schizophrenia. Cochrane Database Syst Rev. 2013;2:CD007253.
179. Centers for Disease Control and Prevention. Cigarette Smoking During Pregnancy: United States, 2016. Available at https://https://www.cdc.gov/nchs/products/databriefs/db305.htm. Last accessed May 3, 2022.
180. Centers for Disease Control and Prevention. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease. A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services; 2010.
181. Dietz PM, England LJ, Shapiro-Mendoza CK, Tong VT, Farr SL, Callaghan WM. Infant morbidity and mortality attributable to prenatal smoking in the United States. Am J Prev Med. 2010;39(1):45-52.
182. Buka SL, Shenassa ED, Niaura R. Elevated risk of tobacco dependence among offspring of mothers who smoked during pregnancy: a 30-year prospective study. Am J Psychiatry. 2003;160(11):1978-1984.
183. Osler M, Clausen J, Ibsen KK, Jensen G. Maternal smoking during childhood and increased risk of smoking in young adulthood.Int J Epidemiol. 1995;24(4):710-714.
184. Centers for Disease Control and Prevention. Smoking and Tobacco Use: Smoking in the Movies. Available at https://www.cdc.gov/tobacco/data_statistics/fact_sheets/youth_data/movies/index.htm Last accessed May 3, 2022.
185. Ohida T, Kaneita Y, Osaki Y, et al. Is passive smoking associated with sleep disturbance among pregnant women? Sleep. 2007;30(9):1155-1161.
186. Mosier HD Jr, Jansons RA. Distribution and fate of nicotine in the rat fetus. Teratology. 1972;6(3):303-311.
187. Suzuki K, Horiguchi T, Comas-Urrutia AC, Mueller-Heubach E, Morishima HO, Adamsons K. Placental transfer and distribution of nicotine in the pregnant rhesus monkey. Am J Obstet Gynecol. 1974;119(2):253-262.
188. Pastrakuljic A, Schwartz R, Simone C, Derewlany LO, Knie B, Koren G. Transplacental transfer and biotransformation studies of nicotine in the human placental cotyledon perfused in vitro. Life Sci. 1998;63(26):2333-2342.
189. Luck W, Nau H, Hansen R, Steldinger R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Dev Pharmacol Ther. 1985;8(6):384-395.
190. Sastry BV. Placental toxicology: tobacco smoke, abused drugs, multiple chemical interactions, and placental function. Reprod Fertil Dev. 1991;3(4):355-372.
191. Demir R, Demir AY, Yinanc M. Structural changes in placental barrier of smoking mother: a quantitative and ultrastructural study. Pathol Res Pract. 1994;190(7):656-667.
193. Klebanoff MA, Levine RJ, Clemens JD, DerSimonian R, Wilkins DG. Serum cotinine concentration and self-reported smoking during pregnancy. Am J Epidemiol. 1998;148(3):259-262.
194. Jauniaux E, Gulbis B, Acharya G, Thiry P, Rodeck C. Maternal tobacco exposure and cotinine levels in fetal fluids in the first half of pregnancy. Obstet Gynecol. 1999;93(1):25-29.
195. Dempsey D, Jacob P III, Benowitz NL. Nicotine metabolism and elimination kinetics in newborns. Clin Pharmacol Ther. 2000;67(5):458-465.
196. Loebstein R, Lalkin A, Koren G. Pharmacokinetic changes during pregnancy and their clinical relevance. Clin Pharmacokinet. 1997;33(5):328-343.
197. Dempsey D, Jacob P III, Benowitz NL. Accelerated metabolism of nicotine and cotinine in pregnant smokers. J Pharmacol Exp Ther. 2002;301(2):594-598.
198. Muneoka K, Ogawa T, Kamei K, et al. Prenatal nicotine exposure affects the development of the central serotonergic system as well as the dopaminergic system in rat offspring: involvement of route of drug administrations. Brain Res Dev Brain Res. 1997;102(1):117-126.
199. Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther. 1998;285(3):931-945.
200. Slotkin TA, Lappi SE, McCook EC, Lorber BA, Seidler FJ. Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull. 1995;38(1):69-75.
201. Kinney HC, O'Donnell TJ, Kriger P, White WS. Early developmental changes in (3H) nicotine binding in the human brainstem. Neuroscience. 1993;55(4):1127-1130.
202. Lewis KW, Bosque EM. Deficient hypoxia awakening response in infants of smoking mothers: possible relationship to sudden infant death syndrome. J Pediatr. 1995;127(5):691-696.
203. Roy TS, Seidler FJ, Slotkin TA. Prenatal nicotine exposure evokes alterations of cell structure in hippocampus and somatosensory cortex. J Pharmacol Exp Ther. 2002;300(1):124-133.
204. Stick S. Pediatric origins of adult lung disease. 1. The contribution of airway development to paediatric and adult lung disease. Thorax. 2000;55(7):587-594.
205. Sekhon HS, Proskocil BJ, Clark JA, Spindel ER. Prenatal nicotine exposure increases connective tissue expression in foetal monkey pulmonary vessels. Eur Respir J. 2004;23(6):906-915.
206. Collins MH, Moessinger AC, Kleinerman J, et al. Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Pediatr Res. 1985;19(4):408-412.
207. Maritz GS, Windvogel S. Chronic maternal nicotine exposure during gestation and lactation and the development of the lung parenchyma in the offspring: response to nicotine withdrawal. Pathophysiology. 2003;10(1):69-75.
208. Economides D, Braithwaite J. Smoking, pregnancy and the fetus. J R Soc Health. 1994;114(4):198-201.
209. Philipp K, Pateisky N, Endler M. Effects of smoking on uteroplacental blood flow. Gynecol Obstet Invest. 1984;17(4):179-182.
210. Tolson CM, Seidler FJ, McCook EC, Slotkin TA. Does concurrent or prior nicotine exposure interact with neonatal hypoxia to produce cardiac cell damage? Teratology. 1995;52(5):298-305.
211. Navarro HA, Mills E, Seidler FJ, et al. Prenatal nicotine exposure impairs beta-adrenergic function: persistent chronotropic subsensitivity despite recovery from deficits in receptor binding. Brain Res Bull. 1990;25(2):233-237.
212. Slotkin TA, Saleh JL, McCook EC, Seidler FJ. Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology. 1997;55(3): 177-184.
213. Hafström O, Milerad J, Sandberg KL, Sundell HW. Cardiorespiratory effects of nicotine exposure during development. Respir Physiol Neurobiol. 2005;149(1-3):325-341.
214. Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42(2):270-283.
215. Sartiani L, Cerbai E, Lonardo G, et al. Prenatal exposure to carbon monoxide affects postnatal cellular electrophysiological maturation of the rat heart: a potential substrate for arrhythmogenesis in infancy. Circulation. 2004;109(3):419-423.
216. Lampl M, Kuzawa CW, Jeanty P. Prenatal smoke exposure alters growth in limb proportions and head shape in the midgestation human fetus. Am J Hum Biol. 2003;15(4):533-546.
217. Lampl M, Kuzawa CW, Jeanty P. Growth patterns of the heart and kidney suggest inter-organ collaboration in facultative fetal growth. Am J Hum Biol. 2005;17(2):178-194.
218. DiFranza JR, Aligne CA, Weitzman M. Prenatal and postnatal environmental tobacco smoke exposure and children's health. Pediatrics. 2004;113(4 Suppl):1007-1015.
219. Simpson WJ. A preliminary report on cigarette smoking and the incidence of prematurity. Am J Obstet Gynecol. 1957;73(4):807-815.
220. Bernstein IM, Mongeon JA, Badger GJ, Solomon L, Heil SH, Higgins ST. Maternal smoking and its association with birth weight. Obstet Gynecol. 2005;106(5 Pt 1):986-991.
221. Martin JA, Hamilton BE, Sutton PD, Ventura SJ, Menacker F, Munson ML. Births: final data for 2002. Natl Vital Stat Rep. 2003;52(10):1-113.
222. Ontario Medical Association. Rethinking stop-smoking medications: treatment myths and medical realities (update 2008). Medical Rev. 2008;75:22-34.
223. Aagaard-Tillery KM, Porter TF, Lane RH, Varner MW, Lacoursiere DY. In utero tobacco exposure is associated with modified effects of maternal factors on fetal growth. Am J Obstet Gynecol. 2008;198(1):66.e1-66.e6.
224. Akalin-Sel T, Campbell S. Understanding the pathophysiology of intra-uterine growth retardation: the role of the "lower limb reflex" in redistribution of blood flow. Eur J Obstet Gynecol Reprod Biol. 1992;46(2-3):79-86.
225. Hammarén-Malmi S, Tarkkanen J, Mattila PS. Analysis of risk factors for childhood persistent middle ear effusion. Acta Otolaryngol. 2005;125(10):1051-1054.
226. Stathis SL, O'Callaghan DM, Williams GM, Najman JM, Andersen MJ, Bor W. Maternal cigarette smoking during pregnancy is an independent predictor for symptoms of middle ear disease at five years' postdelivery. Pediatrics. 1999;104(2):e16.
227. Hecht SS, Hoffmann D. Tobacco-specific nitrosamines, an important group of carcinogens in tobacco and tobacco smoke. Carcinogenesis. 1988;9(6):875-884.
228. Shopland DR (ed). The Health Consequences of Smoking: Cancer: A Report of the Surgeon General. Rockville, MD: U.S. Public Health Service Office on Smoking and Health; 1982.
229. Myers SR, Spinnato JA, Pinorini-Godly MT, Cook C, Boles B, Rodgers GC. Characterization of 4-aminobiphenyl-hemoglobin adducts in maternal and fetal blood-samples. J Toxicol Environ Health. 1996;47(6):553-566.
230. Hofhuis W, de Jongste JC, Merkus PJ. Adverse health effects of prenatal and postnatal tobacco smoke exposure on children. Arch Dis Child. 2003;88(12):1086-1090.
231. Boffetta P, Trédaniel J, Greco A. Risk of childhood cancer and adult lung cancer after childhood exposure to passive smoke: a meta-analysis. Environ Health Perspect. 2000;108(1):73-82.
232. Cooper C, Harvey N, Cole Z, Hanson M, Dennison E. Developmental origins of osteoporosis: the role of maternal nutrition. Adv Exp Med Biol. 2009;646:31-39.
233. Fried PA, Watkinson B. 12- and 24-month neurobehavioural follow-up of children prenatally exposed to marijuana, cigarettes and alcohol. Neurotoxicol Teratol. 1988;10(4):305-313.
234. Kristjansson B, Fried PA, Watkinson B. Maternal smoking during pregnancy affects children's vigilance performance. Drug Alcohol Depend. 1989;24(1):11-19.
235. Naeye RL, Peters EC. Mental development of children whose mothers smoked during pregnancy. Obstet Gynecol. 1984;64(5):601-607.
236. Streissguth AP, Barr HM, Martin DC, Herman CS. Effects of maternal alcohol, nicotine, and caffeine use during pregnancy on infant mental and motor development at eight months. Alcohol Clin Exp Res. 1980;4(2):152-164.
237. Kotimaa AJ, Moilanen I, Taanila A, et al. Maternal smoking and hyperactivity in 8-year-old children. J Am Acad Child Adolesc Psychiatry. 2003;42(7):826-833.
238. Thapar A, Fowler T, Rice F, et al. Maternal smoking during pregnancy and attention deficit hyperactivity disorder symptoms in offspring. Am J Psychiatry. 2003;160(11):1985-1989.
239. Orlebeke JF, Knol DL, Verhulst FC. Child behavior problems increased by maternal smoking during pregnancy. Arch Environ Health. 1999;54(1):15-19.
240. Brook JS, Brook DW, Whiteman M. The influence of maternal smoking during pregnancy on the toddler's negativity. Arch Pediatr Adolesc Med. 2000;154(4):381-385.
241. Brennan PA, Grekin ER, Mednick SA. Maternal smoking during pregnancy and adult male criminal outcomes. Arch Gen Psychiatry. 1999;56(3):215-219.
242. Ferguson DM, Woodward LJ, Horwood LJ. Maternal smoking during pregnancy and psychiatric adjustment in late adolescence. Arch Gen Psychiatry. 1998;55:721-727.
243. Hellström-Lindahl E, Nordberg A. Smoking during pregnancy: a way to transfer the addiction to the next generation? Respiration. 2002;69(4):289-293.
244. Chan-Yeung M, Dimich-Ward H. Respiratory health effects of exposure to environmental tobacco smoke. Respirology. 2003;8(2):131-139.
245. Cook DG, Strachan DP. Health effects of passive smoking-10: summary of effects of parental smoking on the respiratory health of children and implications for research. Thorax. 1999;54(4):357-366.
246. Miller T, Rauh VA, Glied SA, et al. The economic impact of early life environmental tobacco smoke exposure: early intervention for developmental delay. Environ Health Perspect. 2006;114(10):1585-1588.
247. Ueda Y, Stick SM, Hall G, Sly PD. Control of breathing in infants born to smoking mothers. J Pediatr. 1999;135(2 Pt 1):226-232.
248. Samet JM, Yang G. Passive smoking, women and children. In: Samet JM, Yoon SY (eds). Women and the Tobacco Epidemic: Challenges for the 21st Century. Geneva: The World Health Organization in collaboration with the Institute for Global Tobacco Control and the Johns Hopkins School of Public Health; 2001.
249. Johansson A, Hermansson G, Ludvigsson J. How should parents protect their children from environmental tobacco-smoke exposure in the home? Pediatrics. 2004;113(4):e291-e295.
250. Law KL, Stroud LR, LaGasse LL, Niaura R, Liu J, Lester BM. Smoking during pregnancy and newborn neurobehavior. Pediatrics. 2003;111(6 Pt 1):1318-1323.
251. Abou-Donia MB, Abdel-Rahman A, Goldstein LB, et al. Sensorimotor deficits and increased brain nicotinic acetylcholine receptors following exposure to chlorpyrifos and/or nicotine in rats. Arch Toxicol. 2003;77(8):452-458.
252. Gospe SM Jr, Zhou SS, Pinkerton KE. Effects of environmental tobacco smoke exposure in utero and/or postnatally on brain development. Pediatr Res. 1996;39(3):494-498.
253. Navarro HA, Seidler FJ, Schwartz RD, Baker FE, Dobbins SS, Slotkin TA. Prenatal exposure to nicotine impairs nervous system development at a dose which does not affect viability or growth. Brain Res Bull. 1989;23(3):187-192.
254. Slotkin TA. Cholinergic systems in brain development and disruption by neurotoxicants: nicotine, environmental tobacco smoke, organophosphates. Toxicol Appl Pharmacol. 2004;198(2):132-151.
255. Hutchinson S, Glantz S, Zhu BQ, et al. In-utero and neonatal exposure to secondhand smoke causes vascular dysfunction in newborn rats. J Am Coll Cardiol. 1998;32(5):1463-1467.
256. Török J, Gvozdjáková A, Kucharská J, et al. Passive smoking impairs endothelium-dependent relaxation of isolated rabbit arteries. Physiol Res. 2000;49(1):135-141.
257. Kallio K, Jokinen E. Tobacco smoke exposure is associated with attenuated endothelial function in 11-year-old healthy children. Circulation. 2007;115(25):3205-3212.
258. Simko F, Braunova Z, Kucharska J, Bada V, Kyselovic J, Gvozdjakova A. Passive smoking induced hypertrophy of the left ventricle: effect of captopril. Pharmamazie. 1999;54(4):314.
259. Neufeld EJ, Mietus-Snyder M, Beiser AS, Baker AL, Newburger JW. Passive cigarette smoking and reduced HDL cholesterol levels in children with high risk lipid profiles. Circulation. 1997;96(5):1403-1407.
260. Moskowitz WB, Schwartz PF, Schieken RM. Childhood passive smoking, race and coronary artery disease: the MCV Twin Study. Medical College of Virginia. Arch Pediatr Adolesc Med. 1999;153(5):446-453.
261. Feldman J, Shenker IR, Etzel RA, et al. Passive smoking alters lipid profiles in adolescents. Pediatrics. 1991;88(2):259-264.
262. Centers for Disease Control and Prevention. Sudden Unexpected Infant Death and Sudden Infant Death Syndrome: Data and Statistics. Available at https://www.cdc.gov/sids/data.htm. Last accessed May 3, 2022.
263. Klonoff-Cohen HS, Edelstein SL, Lefkowitz ES, et al. The effect of passive smoking and tobacco exposure through breast milk on sudden infant death syndrome. JAMA. 1995;273(10):795-798.
264. Matturri L, Ottaviani G, Lavezzi AM. Maternal smoking and sudden infant death syndrome: epidemiological study related to pathology. Virchows Arch. 2006;449(6):697-706.
265. Zhang K, Wang X. Maternal smoking and increased risk of sudden infant death syndrome: a meta-analysis. Leg Med (Tokyo). 2013;15(3):115-121.
266. Milerad J, Rajs J, Gidlund E. Nicotine and cotinine levels in pericardial fluid in victims of SIDS. Acta Paediatr. 1994;83(1):59-62.
267. Dunn A, Zeise L (eds). Health Effects of Exposure to Environmental Tobacco Smoke. Sacramento, CA: California Environmental Protection Agency, Office of Environmental Health Hazard Assessment; 1997.
268. Tsimoyianis GV, Jacobson MS, Feldman JG, et al. Reduction in pulmonary function and increased frequency of cough associated with passive smoking in teenage athletes. Pediatrics. 1987;80(1):32-36.
269. Lynn WR (ed). The Health Consequences of Involuntary Smoking: A Report of the Surgeon General, 1986. Rockville, MD: United States Public Health Service Office on Smoking and Health; 1986.
270. U.S. Surgeon General's Advisory Committee on Smoking and Health. Smoking and Health. Report of the Advisory Committee to the Surgeon General of the Public Health Service, 1964. Washington, DC: U.S. Department of Health, Education, and Welfare; 1964.
271. Hanrahan JP, Tager IB, Segal MR, et al. The effect of maternal smoking during pregnancy on early infant lung function. Am Rev Respir Dis. 1992;145(5):1129-1135.
272. Chhabra D, Sharma S, Kho AT, et al. Fetal lung and placental methylation is associated with in utero nicotine exposure. Epigenetics. 2014;9(11):1473-1484.
273. Etzel RA. How environmental exposures influence the development and exacerbation of asthma. Pediatrics. 2003;112(1 Pt 2):233-239.
274. Institute of Medicine Committee on the Assessment of Asthma and Indoor Air. Clearing the Air: Asthma and Indoor Air Exposures. Washington, DC: National Academy Press; 2000.
275. Spahn JD, Szefler SJ. The etiology and control of bronchial hyperresponsiveness in children. Curr Opin Pediatr. 1996;8(6):591-596.
277. Karadag B, Karakoç F, Ceran O, Ersu R, Inan S, Dagli E. Does passive smoke exposure trigger acute asthma attack in children? Allergol Immunopathol (Madr). 2003;31(6):318-323.
278. Aligne CA, Moss ME, Auinger P, Weitzman M. Association of pediatric dental caries with passive smoking. JAMA. 2003;289(10):1258-1264.
279. Wilson KM, Finkelstein JN, Blumkin AK, Best D, Klein JD. Micronutrient levels in children exposed to secondhand tobacco smoke. Nicotine Tob Res. 2011;13(9):800-808.
280. David GL, Koh WP, Lee HP, Yu MC, London SJ. Childhood exposure to environmental tobacco smoke and chronic respiratory symptoms in non-smoking adults: the Singapore Chinese Health Study. Thorax. 2005;60(12):1052-1058.
281. Sandler DP, Everson RB, Wilcox AJ, Browder JP. Cancer risk in adulthood from early life exposure to parents' smoking. Am J Public Health. 1985;75(5):487.
282. Peppone LJ, Piazza KM, Mahoney MC, et al. Associations between adult and childhood secondhand smoke exposures and fecundity and fetal loss among women who visited a cancer hospital. Tob Control. 2009;18(2):115-120.
283. Strohsnitter WC, Hatch EE, Hyer M, et al. The association between in utero cigarette smoke exposure and age at menopause. Am J Epidmiol. 2008;167(6):727-733.
284. Collishaw NE, Kirkbride J, Wigle DT. Tobacco smoke in the workplace: an occupational health hazard. Can Med Assoc J. 1984;131(10):1199-1204.
285. Navas-Acien A, Peruga A, Breysse P, et al. Secondhand tobacco smoke in public places in Latin America, 2002–2003. JAMA. 2004;291(22):2741-2745.
286. Castellan RM, Chosewood LC, Trout D, et al. Current Intelligence Bulletin 67: Promoting Health and Preventing Disease and Injury Through Workplace Tobacco Policies. Morgantown, WV: National Institute for Occupational Safety and Health; 2015.
287. Samet JM, Spengler JD. Indoor environments and health: moving into the 21st century. Am J Public Health. 2003;93(9):1489-1493.
288. Fichtenberg CM, Glantz SA. Effect of smoke-free workplaces on smoking behavior: systematic review. BMC. 2002;325:188-191.
289. Bauer JE, Hyland A, Li Q, Steger C, Cummings KM. A longitudinal assessment of the impact of smoke-free worksite policies on tobacco use. Am J Public Health. 2005;95(6):1024-1029.
290. International Agency for Research on Cancer. Evaluating the effectiveness of smoke-free polices. In: IARC Handbooks of Cancer Prevention, Tobacco Control. Vol. 13. Lyon: World Health Organization, International Agency for Research on Cancer; 2009.
291. Hopkins DP, Razi S, Leeks KD, Priya Kalra G, Chattopadhyay SK, Soler RE. Smokefree policies to reduce tobacco use: a systematic review. Am J Prev Med. 2010;38(2 Suppl):S275-S289.
292. Callinan JE, Clarke A, Doherty K, Kelleher C. Legislative smoking bans for reducing secondhand smoke exposure, smoking prevalence and tobacco consumption. Cochrane Database Syst Rev. 2010;4:CD005992.
293. Eisner MD, Smith AK, Blanc PD. Bartenders' respiratory health after establishment of smokefree bars and taverns. JAMA. 1998;280(22):1909-1914.
294. Lambert WE, Samet JM, Spengler JD. Environmental tobacco smoke concentrations in no-smoking and smoking sections of restaurants. Am J Public Health. 1993;83(9):1339-1341.
295. Chang C, Leighton J, Mostashari F, McCord C, Frieden TR. The New York City Smoke-free Air Act: second-hand smoke as a worker health and safety issue. Am J Ind Med. 2004;46(2):188-195.
296. Fujishiro K, Hinckley Stukovsky KD, Diez Roux A, Landsbergis P, Burchfiel C. Occupational gradients in smoking behavior and exposure to workplace environmental tobacco smoke: The Multi-Ethnic Study of Atherosclerosis (MESA). J Occup Environ Med. 2012;54(2):136-145.
297. Repace J. Flying the smoky skies: secondhand smoke exposure of flight attendants. Tob Control. 2004;13(Suppl 1):i8-i19.
298. Enstrom JE, Kabat GC. Environmental tobacco smoke and coronary heart disease mortality in the United States: a meta-analysis and critique. Inhal Toxicol. 2006;18(3):199-210.
299. Glantz SA, Parmley WW. Passive smoking and heart disease: epidemiology, physiology, and biochemistry. Circulation. 1991;83(1): 1-12.
300. Institute of Medicine Committee on Secondhand Smoke Exposure and Acute Coronary Events. Secondhand Smoke Exposure and Cardiovascular Effects: Making Sense of the Evidence. Washington, DC: National Academies Press; 2010.
302. Sargent RP, Shepard RM, Glantz SA. Reduced incidence of admissions for myocardial infarction associated with public smoking ban: before and after study. BMJ. 2004;328(7446):977-980.
304. Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. 2022;145:e153-e639.
306. Abraham D, Distler O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9(Suppl 2):S2.
307. Okamoto Y, Kihara S, Ouchi N, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106(22):2767-2770.
308. U.S. Department of Health and Human Services. Reducing the Health Consequences of Smoking: 25 Years of Progress. A Report of the Surgeon General: 1989 Executive Summary. Washington, D.C.: Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 1989.
309. Whincup PH, Gilg JA, Emberson JR, et al. Passive smoking and risk of coronary heart disease and stroke: prospective study with cotinine measurement. BMJ. 2004;329(7459):200-205.
310. U.S. Department of Health and Human Services. The Health Benefits of Smoking Cessation. A Report of the Surgeon General: 1990. Rockville, MD: U.S. Public Health Service, Office on Smoking and Health; 1990.
311. Panagiotakos DB, Pitsavos C, Chrysohoou C, et al. Effect of exposure to secondhand smoke on markers of inflammation: the ATTICA study. Am J Med. 2004;116(3):145-150.
312. Glantz SA, Parmley WW. Passive smoking and heart disease: mechanisms and risk. JAMA. 1995;273(13):1047-1053.
313. He J, Vupputuri S, Allen K, Prerost MR, Hughes J, Whelton PK. Passive smoking and the risk of coronary heart disease—a meta-analysis of epidemiologic studies. N Engl J Med. 1999;340(12):920-926.
314. Kawachi I, Colditz GA, Speizer FE, et al. A prospective study of passive smoking and coronary heart disease. Circulation. 1997;95(10):2374-2379.
315. Valkonen M, Kuusi T. Passive smoking induces atherogenic changes in low-density lipoprotein. Circulation. 1998;97(20):2012-2016.
316. Burghuber OC, Punzengruber C, Sinzinger H, Haber P, Silberbauer K. Platelet sensitivity to prostacyclin in smokers and non-smokers. Chest. 1986;90(1):34-38.
317. Benowitz NL, Fitzgerald GA, Wilson M, Zhang Q. Nicotine effects on eicosanoid formation and hemostatic function: comparison of transdermal nicotine and cigarette smoking. J Am Coll Cardiol. 1993;22(4):1159-1167.
318. California Environmental Protection Agency, Office of Environmental Health Hazard Assessment. National Cancer Institute, Smoking and Tobacco Control Monographs. Monograph 10: Health Effects of Exposure to Environmental Tobacco Smoke. Available at https://cancercontrol.cancer.gov/brp/tcrb/monographs/10/index.html. Last accessed May 3, 2022.
319. Scientific Committee on Tobacco and Health. Report of the Scientific Committee on Tobacco and Health. Available at https://www.gov.uk/government/publications/report-of-the-scientific-committee-on-tobacco-and-health. Last accessed May 2, 2022.
320. Taylor AE, Johnson DC, Kazemi H. Environmental tobacco smoke and cardiovascular disease: a position paper from the Council on Cardiopulmonary and Critical Care, American Heart Association. Circulation. 1992;86(2):699-702.
321. Glymour MM, Defries TB, Kawachi I, Avendano M. Spousal smoking and incidence of first stroke: the Health and Retirement Study. Am J Prev Med. 2008;35(3):245-248.
322. Janson C. The effect of passive smoking on respiratory health in children and adults. Int J Tuberc Lung Dis. 2004;8(5):510-516.
323. Leuenberger P, Schwartz J, Ackerman-Liebrich U, et al. Passive smoking exposure in adults and chronic respiratory symptoms (SAPALDIA study). Am J Respir Crit Care Med. 1994;150(5 Pt 1):1222-1228.
324. Eisner MD, Balmes J, Katz PP, Trupin L, Yelin EH, Blanc PD. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environ Health. 2005;4(1):7.
325. Schick S, Glantz S. Philip Morris toxicological experiments with fresh sidestream smoke: more toxic than mainstream smoke. Tob Control. 2005;14(6):396-404.
326. Jaakkola MS, Piipari R, Jaakkola N, Jaakkola JJ. Environmental tobacco smoke and adult-onset asthma: a population-based incident case-control study. Am J Public Health. 2003;93(12):2055-2060.
327. Dahms TE, Bolin JF, Slavin RG. Passive smoking: effects on bronchial asthma. Chest. 1981;80(5):530-534.
328. Zhong L, Goldberg MS, Gao YT, Jin F. A case-control study of lung cancer and environmental tobacco smoke among nonsmoking women living in Shanghai, China. Cancer Causes Control. 1999;10(6):607-616.
329. Murphy TD. Passive Smoking and Lung Disease. Available at https://emedicine.medscape.com/article/1005579-overview. Last accessed May 3, 2022.
330. Khuder SA. Effect of cigarette smoking on major histological types of lung cancer: a meta-analysis. Lung Cancer. 2001;31(2-3):139-148.
331. Gentzke AS, Wang TW, Cornelius M, et al. Tobacco product use and associated factors among middle and high school students— National Youth Tobacco Survey, United States, 2021. MMWR Surveill Summ. 2022;71(No. SS-5):1–29.
332. Ketterer B, Harris JM, Talaska G, et al. The human glutathione S-transferase supergene family, its polymorphism, and its effects on susceptibility to lung cancer. Environ Health Perspect. 1992;98:87-94.
333. Kihara M, Kihara M, Noda K. Risk of smoking for squamous and small cell carcinomas of the lung modulated by combinations of CYP1A1 and GSTM1 gene polymorphisms in a Japanese population. Carcinogenesis. 1995;16(10):2331-2336.
334. McWilliams JE, Sanderson BJ, Harris EL, Richert-Boe KE, Henner WD. Glutathione S-transferase M1 (GSTM1) deficiency and lung cancer risk. Cancer Epidemiol Biomarkers Prev. 1995;4(6):589-594.
335. Saarikoski ST, Voho A, Reinikainen M, et al. Combined effect of polymorphic GST genes on individual susceptibility to lung cancer. Int J Cancer. 1998;77(4):516-521.
336. Seidegård J, Vorachek WR, Pero RW, Pearson WR. Hereditary differences in the expression of the human glutathione transferase active on trans-stilbene oxide are due to a gene deletion. Proc Natl Acad Sci USA. 1988;85(19):7293-7297.
337. Bennett WP, Alavanja MC, Blomeke B, et al. Environmental tobacco smoke, genetic susceptibility, and risk of lung cancer in never-smoking women. J Natl Cancer Inst. 1999;91(23):2009-2014.
338. Rebbeck TR. Molecular epidemiology of the human glutathione S-transferase genotypes GSTM1 and GSTT1 in cancer susceptibility. Cancer Epidemiol Biomarkers Prev. 1997;6(9):733-743.
339. Tang DL, Rundle A, Warburton D, et al. Associations between both genetic and environmental biomarkers and lung cancer: evidence of a greater risk of lung cancer in women smokers. Carcinogenesis. 1998;19(11):1949-1953.
340. Kelsey KT, Spitz MR, Zuo ZF, Wiencke JK. Polymorphisms in the glutathione S-transferase class mu and theta genes interact and increase susceptibility to lung cancer in minority populations (Texas, United States). Cancer Causes Control. 1997;8(4):554-559.
341. Kawajiri K, Nakachi K, Imai K, Watanabe J, Hayashi S. The CYP1A1 gene and cancer susceptibility. Crit Rev Oncol Hematol. 1993;14(1):77-87.
342. Kawajiri K, Nakachi K, Imai K, Hayashi S, Watanabe J. Individual differences in lung cancer susceptibility in relation to polymorphisms of P-450IA1 gene and cigarette dose. Princess Takamatsu Symp. 1990;21:55-61.
343. Yokota J, Shiraishi K, Kohno T. Genetic basis for susceptibility to lung cancer: recent progress and future directions. Adv Cancer Res. 2010;109:51-72.
344. Trunog T, Hung RJ, Amos CI, et al. Replication of lung cancer susceptibility loci at chromosomes 15q25, 5p15, and 6p21: a pooled analysis from the International Lung Cancer Consortium. J Natl Cancer Inst. 2010;102(13):959-971.
345. Walsh KM, Gorlov IP, Hansen HM et al. Fine-mapping of the 5p15.33, 6p22.1-p21.31, and 15q25.1 regions identifies functional and histology-specific lung cancer susceptibility loci in African-Americans. Cancer Epidemiol Biomarkers Prev. 2013;22(2):251-260.
346. Houston TK, Person SD, Pletcher MJ, Liu K, Iribarren C, Kiefe CI. Active and passive smoking and development of glucose intolerance among young adults in a prospective cohort: CARDIA study. BMJ. 2006;332(7549):1064-1069.
347. Pan A, Wang Y, Talaei M, Hu FB, Wu T. Relation of active, passive, and quitting smoking with incident type 2 diabetes: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2015;3(12):958-967.
348. Setty AR, Curhan G, Choi HK. Smoking and the risk of psoriasis in women: Nurses' Health Study II. Am J Med. 2007;120(11):953-959.
349. Khan JC, Thurlby DA, Shahid H, et al. Smoking and age-related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br J Ophthalmol. 2006;90(1):75-80.
350. World Health Organization. Cervical Cancer. Available at https://www.who.int/health-topics/cervical-cancer. Last accessed May 3, 2022.
352. Messina ES, Tyndale RF, Sellers EM. A major role for CYP2A6 in nicotine C-oxidation by human liver microsomes. J Pharmacol Exp Ther. 1997;282(3):1608-1614.
353. Benowitz NL, Jacob P III. Metabolism of nicotine to cotinine studied by a dual stable isotope method. Clin Pharmacol Ther. 1994;56(5):483-493.
354. Lambers DS, Clark KE. The maternal and fetal physiologic effects of nicotine. Semin Perinatol. 1996;20(2):115-126.
355. Benowitz NL. Biomarkers of environmental tobacco smoke exposure. Environ Health Perspect. 1999;107(Suppl 2):349-355.
356. Mulcahy M, Evans DS, Hammond SK, Repace JL, Byrne M. Secondhand smoke exposure and risk following the Irish smoking ban: an assessment of salivary cotinine concentrations in hotel workers and air nicotine levels in bars. Tob Control. 2005;14(6):384-388.
357. Gordon SM, Wallace LA, Brinkman MC, Callahan PJ, Kenny DV. Volatile organic compounds as breath biomarkers for active and passive smoking. Environ Health Perspect. 2002;110(7):689-698.
358. Agency for Toxic Substances and Disease Registry. Draft of Toxicological Profile for 1, 3-butadiene. Available at https://www.atsdr.cdc.gov/toxprofiles/tp28-c1.pdf. Last accessed May 3, 2022.
360. Husgafvel-Pursiainen K, Boffetta P, Kannio A, et al. p53 mutations and exposure to environmental tobacco smoke in a multicenter study on lung cancer. Cancer Res. 2000;60(11):2906-2911.
361. Hovell MF, Zakarian JM, Wahlgren DR, Matt GE, Emmons KM. Reported measures of environmental tobacco smoke exposure: trials and tribulations. Tob Control. 2000;9(Suppl 3):III22-III28.
362. Haufroid V, Lison D. Urinary cotinine as a tobacco-smoke exposure index: a minireview. Int Arch Occup Environ Health. 1998;71(3):162-168.
363. Winickoff JP, Friebely J, Tanski SE, et al. Beliefs about the health effects of "thirdhand" smoke and home smoking bans. Pediatrics. 2009;123:e74-e79.
364. Hein HO, Suadicani P, Skov P, Gyntelberg F. Indoor dust exposure: an unnoticed aspect of involuntary smoking. Arch Environ Health. 1991;46(2):98-101.
365. Matt GE, Quintana PJ, Hovell MF, et al. Households contaminated by environmental tobacco smoke: sources of infant exposures. Tob Control. 2004;13(1):29-37.
366. Al-Delaimy WK, Crane J, Woodward A. Passive smoking in children: effect of avoidance strategies, at home as measured by hair nicotine levels. Arch Environ Health. 2001;56(2):117-122.
367. Bonanno LJ, Freeman NCG, Greenberg M, Lioy PJ. Multivariate analysis on levels of selected metals, particulate matter, VOC, and household characteristics and activities from the midwestern states NHEXAS. Appl Occup Environ Hyg. 2001;16(9): 859-874.
368. Smith CJ, Perfetti TA, Garg R, Martin P, Hansch C. Percutaneous penetration enhancers in cigarette mainstream smoke. Food Chem Toxicol. 2004;42(1):9-15.
369. Schick SF, Glantz S. Concentrations of the carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in sidestream cigarette smoke increase after release into indoor air: results from unpublished tobacco industry research. Cancer Epidemiol Biomarkers Prev. 2007;16(8):1547-1553.
370. Ghosh D, Mishra MK, Das S, Kaushik DK, Basu A. Tobacco carcinogen induces microglial activation and subsequent neuronal damage. J Neurochem. 2009;110(3):1070-1081.
371. Oie L, Nafstad P, Botten G, Magnus O, Jaakkola JK. Ventilation in homes and bronchial obstruction in young children. Epidemiology. 1999;10(3):294-299.
372. Matt GE, Quintana PJ, Hovell MF, et al. Residual tobacco smoke pollution in used cars for sale: air, dust, and surfaces. Nicotine Tob Res. 2008;10(9):1467-1475.
373. Haussmann HJ, Anskeit E, Becker D, et al. Comparison of fresh and room-aged cigarette sidestream smoke in a subchronic inhalation study on rats. Toxicol Sci. 1998;41(1):100-116.
374. Rao SP, Sikora L, Hosseinkhani MR, Pinkerton KE, Sriramarao P. Exposure to environmental tobacco smoke induces angiogenesis and leukocyte trafficking in lung microvessels. Exp Lung Res. 2009;35(2):119-135.
375. Schick S, Glantz SA. Sidestream cigarette smoke toxicity increases with aging and exposure duration. Tob Control. 2006;15(6):424-429.
376. Godoy P, Castilla J, Mayoral JM, et al. Smoking may increase the risk of influenza hospitalization and reduce influenza vaccine effectiveness in the elderly. Eur J Public Health. 2018;28(1):150-155.
377. Centers for Disease Control and Prevention. Quitting smoking among adults—United States, 2000–2015. MMWR. 2017;65(52):1457-1464.
378. Jorenby DE, Fiore MC. The Agency for Health Care Policy and Research smoking cessation clinical practice guideline: basics and beyond. Prim Care. 1999;26(3):513-528.
379. Heatherton TF, Kozlowski LT, Frecker RC, Fagerström KO. The Fagerström Test for nicotine dependence: a revision of the Fagerström Tolerance Questionnaire. Br J Addict. 1991;86(9):1119-1127.
380. Prochaska JO, DiClemente CC, Norcross JC. In search of how people change: applications to addictive behaviors. Am Psychol. 1992;47(9):1102-1114.
381. Marlow SP, Stoller JK. Smoking cessation. Respir Care. 2003;48(12):1238-1254; discussion 1254-1256.
382. Milch CE, Edmunson JM, Beshansky JR, Griffith JL, Selker HP. Smoking cessation in primary care: a clinical effectiveness trial of two simple interventions. Prev Med. 2004;38(3):284-294.
383. Smith PM, Burgess E. Smoking cessation initiated during hospital stay for patients with coronary artery disease: a randomized controlled trial. CMAJ. 2009;180(13):1297-1303.
384. Fiore MC, Jaen CR, Baker TB, et al. Treating Tobacco Use and Dependence: 2008 Update: Clinical Practice Guideline. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service; 2008.
385. Mullen PD, Carbonari JP, Tabak ER, Glenday MC. Improving disclosure of smoking by pregnant women. Am J Obstet Gynecol. 1991;165(2):409-413.
386. Ahluwalia JS, Gibson CA, Kenney RE, Wallace DD, Resnicow K. Smoking status as a vital sign. J Gen Intern Med. 1999;14(7):402-408.
387. Doescher MP, Saver BG. Physicians' advice to quit smoking: the glass remains half empty. J Fam Pract. 2000;49(6):543-547.
388. Brandon TH. Behavioral tobacco cessation treatments: yesterday's news or tomorrow's headlines? J Clin Oncol. 2001;19(18 Suppl):64S-68S.
389. Miller WR. Motivational interviewing with problem drinkers. Behavioural Psychotherapy. 1983;11:147-172.
390. Lai DT, Cahill K, Qin Y, Tang, JL. Motivational interviewing for smoking cessation. Cochrane Database Syst Rev. 2010;(1):CD006936.
391. Lindson-Hawley N, Thompson TP, Begh R. Motivational interviewing for smoking cessation. Cochrane Database Syst Rev. 2015;3:CD006936.
392. Ruger JP, Weinstein MC, Hammond SK, Kearney MH, Emmons KM. Cost-effectiveness of motivational interviewing for smoking cessation and relapse prevention among low-income pregnant women: a randomized controlled trial. Value Health. 2008;11(2):191-198.
393. Lee E. Cross-cultural communication: therapeutic use of interpreters. In: Lee E (ed). Working with Asian Americans: A Guide for Clinicians. New York, NY: The Guilford Press; 1997: 477-489.
394. Thompson RS, Michnich ME, Friedlander L, Gilson B, Grothaus LC, Storer B. Effectiveness of smoking cessation interventions integrated into primary care practice. Med Care. 1988;26(1):62-76.
395. Maciosek MV, Coffield AB, Flottemesch TJ, Edwards NM, Solberg LI. Greater use of preventive services in U.S. health care could save lives at little or no cost. Health Aff (Milwood). 2010;29(9):1656-1660.
396. Cromwell J, Bartosch WJ, Fiore MC, Hasselblad V, Baker T. Cost-effectiveness of the clinical practice recommendations in the AHCPR guideline for smoking cessation. JAMA. 1997;278(21):1759-1766.
397. Hall S, Rugg D, Tunstall C, Jones RT. Preventing relapse to cigarette smoking by behavioral skill training. J Consult Clin Psychol. 1984;52(3):372-382.
398. Stevens VJ, Hollis JF. Preventing smoking relapse using an individually tailored skills-training technique. J Consult Clin Psychol. 1989;57(3):420-424.
399. Tiffany ST, Martin EM, Baker TB. Treatments for cigarette smoking: an evaluation of the contributions of aversion and counseling procedures. Behav Res Ther. 1986;24(4):437-452.
400. Hajek P, Stead LF. Aversive smoking for smoking cessation. Cochrane Database Syst Rev. 2004;(3):CD000546.
401. Hajek P, Stead LF. Aversive smoking for smoking cessation. Cochrane Database Syst Rev. 2000;(2):CD000546.
402. Cinciripini PM, Lapitsky LG, Wallfisch A, Mace R, Nezami E, Van Vunakis H. An evaluation of a multicomponent treatment program involving scheduled smoking and relapse prevention procedures: initial findings. Addict Behav. 1994;19(1):13-22.
403. Pfizer Inc. Chantix (Varenicline) Tablets. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021928s008lbl.pdf. Last accessed May 3, 2022.
404. Stead LF, Perera R, Bullen C, et al. Nicotine replacement therapy for smoking cessation. Cochrane Database Syst Rev. 2012:CD000146.
405. Gorsline J, Okerholm RA, Rolf CN, Moos CD, Hwang SS. Comparison of plasma nicotine concentrations after application of nicoderm (nicotine transdermal system) to different skin sites. J Clin Pharmacol. 1992;32(6):576-581.
406. Orleans CT, Resch N, Noll E, et al. Use of transdermal nicotine in a state-level prescription plan for the elderly: a first look at "real-world" patch users. JAMA. 1994;271:601-607.
408. Schnoll RA, Patterson F, Wileyto EP, et al. Effectiveness of extended-duration transdermal nicotine therapy: a randomized trial. Ann Intern Med. 2010;152(3):144-151.
409. Schnoll RA, Goelz PM, Veluz-Wilkins A, et al. Long-term nicotine replacement therapy: a randomized clinical trial. JAMA Intern Med. 2015;175(4):504-511.
410. Frishman WH, Ky T, Ismail A. Tobacco smoking, nicotine, and nicotine and non-nicotine replacement therapies. Heart Dis. 2001;3(6):365-377.
411. Schneider NG, Jarvik ME, Forsythe AB, Read LL, Elliott ML, Schweiger A. Nicotine gum in smoking cessation: a placebo-controlled, double-blind trial. Addict Behav. 1983;8(3):253-261.
412. Shiffman S, Di Marino ME, Pillitteri JL. The effectiveness of nicotine patch and nicotine lozenge in very heavy smokers. J Subst Abuse Treat. 2005;28(1):49-55.
413. Hurt RD. New medications for nicotine dependence treatment. Nicotine Tob Res. 1999;1(Suppl 2):S175-S179, S207-S210.
414. Molander L, Lunell E, Fagerström KO. Reduction of tobacco withdrawal symptoms with a sublingual nicotine tablet: a placebo-controlled study. Nicotine Tob Res. 2000;2(2):187-191.
415. Tønnesen P, Mikkelsen K, Bremann L. Nurse-conducted smoking cessation in patients with COPD using nicotine sublingual tablets and behavioral support. Chest. 2006;130(2):334-342.
416. Sun H, Guo S, Chen DF, et al. Family support and employment as predictors of smoking cessation success: a randomized, double-blind, placebo-controlled trial of nicotine sublingual tablets in Chinese smokers. Am J Drug Alcohol Abuse. 2009;35(3):183-188.
417. Molander L, Lunell E. Pharmacokinetic investigation of a nicotine sublingual tablet. Eur J Clin Pharmacol. 2001;56(11):813-819.
418. Shiffman S. Effect of nicotine lozenges on affective smoking withdrawal symptoms: secondary analysis of a randomized, double-blind, placebo-controlled clinical trial. Clin Ther. 2008;30(8):1461-1475.
419. Hjalmarson A, Franzon M, Westin A, Wiklund O. Effect of nicotine nasal spray on smoking cessation: a randomized, placebo-controlled, double-blind study. Arch Intern Med. 1994;154(22):2567-2572.
420. Schneider NG, Olmstead R, Mody FV, et al. Efficacy of a nicotine nasal spray in smoking cessation: a placebo-controlled, double-blind trial. Addiction. 1995;90(12):1671-1682.
421. Sutherland G, Stapleton JA, Russell MA, et al. Randomised controlled trial of nasal nicotine spray in smoking cessation. Lancet. 1992;340(8815):324-329.
422. Silagy C, Mant D, Fowler G, Lodge M. Meta-analysis on efficacy of nicotine replacement therapies in smoking cessation. Lancet. 1994;343(8890):139-142.
423. Schneider NG, Lunell E, Olmstead RE, Fagerström KO. Clinical pharmacokinetics of nasal nicotine delivery: a review and comparison to other nicotine systems. Clin Pharmacokinet. 1996;31(1):65-80.
424. Schneider NG, Olmstead RE, Franzon MA, Lunell E. The nicotine inhaler: clinical pharmacokinetics and comparison with other nicotine treatments. Clin Pharmacokinet. 2001;40(9):661-684.
425. Rose JE, Behm FM, Westman EC, Johnson M. Dissociating nicotine and nonnicotine components of cigarette smoking. Pharmacol Biochem Behav. 2000;67(1):71-81.
426. Ascher JA, Cole JO, Colin JN, et al. Bupropion: a review of its mechanism of antidepressant activity. J Clin Psychiatry. 1995;56(9):395-401.
427. Hughes JR, Goldstein MG, Hurt RD, Shiffman S. Recent advances in the pharmacotherapy of smoking. JAMA. 1999;281(1):72-76.
428. Hurt RD, Sachs DP, Glover ED, et al. A comparison of sustained-release bupropion and placebo for smoking cessation. N Engl J Med.1997;337(17):1195-1202.
429. Fiscella K, Franks P. Cost-effectiveness of the transdermal nicotine patch as an adjunct to physicians' smoking cessation counseling. JAMA. 1996;275(16):1247-1251.
430. Lexicomp Online. Available at https://online.lexi.com. Last accessed May 3, 2022.
431. Tonstad S, Tønnesen P, Hajek P, et al. Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA. 2006;296(1):64-71.
432. Gonzales D, Rennard SI, Nides M, et al. Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs. sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA. 2006;296(1):47-55.
433. Jorenby DE, Hays JT, Rigotti NA, et al. Efficacy of varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs. placebo or sustained-release bupropion for smoking cessation: a randomized controlled trial. JAMA. 2006;296(1):56-63.
434. Mills EJ, Wu P, Lockhart I, Thorlund K, Puhan M, Ebbert JO. Comparisons of high-dose and combination nicotine replacement therapy, varenicline, and bupropion for smoking cessation: a systematic review and multiple treatment meta-analysis. Ann Med. 2012;44(6):588-597.
435. Chang PH, Chiang CH, Ho WC, Wu PZ, Tsai JS, Guo FR. Combination therapy of varenicline with nicotine replacement therapy is better than varenicline alone: a systematic review and meta-analysis of randomized controlled trials. BMC Public Health. 2015;22:689.
436. Gourlay SG, Stead LF, Benowitz NL. Clonidine for smoking cessation. Cochrane Database Syst Rev. 2000;(2):CD000058.
437. Lancaster T, Stead LF. Silver acetate for smoking cessation. Cochrane Database Syst Rev. 2000;(2):CD000191.
438. Lancaster T, Stead LF. Silver acetate for smoking cessation. Cochrane Database Syst Rev. 2012;9:CD000191.
439. Lancaster T, Stead LF. Mecamylamine (a nicotine antagonist) for smoking cessation (review). Cochrane Database Syst Rev. 2000;(2):CD001009.
440. Rose JE, Behm FM, Westman EC, Levin ED, Stein RM, Ripka GV. Mecamylamine combined with nicotine skin patch facilitates smoking cessation beyond nicotine patch treatment alone. Clin Pharmacol Ther. 1994;56(1):86-99.
442. Fagerström K. New perspectives in the treatment of tobacco dependence. Monaldi Arch Chest Dis. 2003;60(3):179-183.
443. National Institute on Drug Abuse. Medication Reduces Metabolism of Nicotine, Decreasing Urge to Smoke. Available at https://archives.drugabuse.gov/news-events/news-releases/2000/07/medication-reduces-metabolism-nicotine-decreasing-urge-to-smoke. Last accessed May 3, 2022.
444. Zhang W, Kilicarslan T, Tyndale RF, Sellers EM. Evaluation of methoxsalen, tranylcypromine, and tryptamine as specific and selective CYP2A6 inhibitors in vitro. Drug Metab Dispos. 2001;26(6):897-902.
445. Hughes JR, Higgins ST, Bickel WK. Nicotine withdrawal versus other drug withdrawal syndromes: similarities and dissimilarities. Addiction. 1994;89(11):1461-1470.
446. Centers for Disease Control and Prevention. Current cigarette smoking among adults—United States, 2016. MMWR. 2018;67(2);54-59.
447. Josefson D. U.S. flight attendants win settlement over passive smoking. BMJ. 1997;315(7114):968.
448. Flight Attendant Medical Research Institute. Mission Statement. Available at https://famri.org/mission-statement. Last accessed May 3, 2022.
449. Henningfield JE, Benowitz NL, Connolly GN, et al. Reducing tobacco addiction through tobacco product regulation. Tob Control. 2004;13(2):132-135.
450. Campaign for Tobacco-Free Kids. Voters Across the Country Support Significant Increases in Cigarette Taxes. Available at https://www.tobaccofreekids.org/research/factsheets/0026.pdf. Last accessed May 3, 2022.
451. Campaign for Tobacco-Free Kids. Raising Tobacco Taxes: A Win-Win-Win. Available at https://www.tobaccofreekids.org/assets/factsheets/0385.pdf. Last accessed May 3, 2022.
452. Centers for Disease Control and Prevention. QuickStats: percentage of births to mothers who reported smoking cigarettes at any time during pregnancy, by urbanization level of county of residence—United States, 2020. MMWR. 2021;70:1652.
453. Campaign for Tobacco-Free Kids. State Excise and Sales Taxes Per Pack of Cigarettes Total Amounts and State Rankings. Available at https://www.tobaccofreekids.org/assets/factsheets/0202.pdf. Last accessed May 3, 2022.
454. Eriksen MP, Gottlieb NH. A review of the health impact of smoking control at the workplace. Am J Health Promot. 1998;13(2):83-104.
455. Green E, Courage C, Rushton L. Reducing domestic exposure to environmental tobacco smoke: a review of attitudes and behaviours. J R Soc Promot Health. 2003;123(1):46-51.
456. Cornelius ME, Loretan CG, Wang TW, Jamal A, Homa DM. Tobacco product use among adults—United States, 2020. MMWR. 2022;71(11):397-405.
457. National Institute on Drug Abuse. DrugFacts: Vaping Devices (Electronic Cigarettes). Available at https://nida.nih.gov/publications/drugfacts/vaping-devices-electronic-cigarettes. Last accessed May 3, 2022.
458. U.S. Food and Drug Administration. FDA's Deeming Regulations for E-Cigarettes, Cigars, and All Other Tobacco Products. Available at https://www.fda.gov/tobaccoproducts/labeling/rulesregulationsguidance/ucm394909.htm. Last accessed May 3, 2022.
459. Singh T, Arrazola RA, Corey CG, et al. Tobacco use among middle and high school students—United States, 2011–2015. MMWR. 2016;65(14):361-367.
460. National Center for Health Statistics. Early Release of Selected Estimates Based on Data from January–September 2018 National Health Interview Survey. Available at https://www.cdc.gov/nchs/nhis/releases/released201903.htm. Last accessed May 3, 2022.
461. Lindson-Hawley N, Banting M, West R, Michie S, Shinkins B, Aveyard P. Gradual versus abrupt smoking cessation: a randomized, controlled noninferiority trial. Ann Intern Med. 2016;164(9):585-592.
462. Scott-Sheldon LA, Lantini R, Jennings EG, et al. Text messaging-based interventions for smoking cessation: a systematic review and meta-analysis. JMIR. 2016;4(2).
463. Baker TB, Piper ME, Stein JH, et al. Effects of nicotine patch vs. varenicline vs combination nicotine replacement therapy on smoking cessation at 26 weeks: a randomized clinical trial. JAMA. 2016;315(4):371-379.
464. U.S. Food and Drug Administration. FDA Takes New Steps to Address Epidemic of Youth E-Cigarette Use, Including a Historic Action. Available at https://www.fda.gov/news-events/press-announcements/fda-takes-new-steps-address-epidemic-youth-e-cigarette-use-including-historic-action-against-more. Last accessed May 3, 2022.
465. Leone FT, Zhang Y, Evers-Casey S, et al. Initiating pharmacologic treatment in tobacco-dependent adults: an official American Thoracic Society (ATS) clinical practice guideline. Am J Respir Crit Care Med. 2020;202(2):e5-e31.
466. Heger A, Sator M, Walch K, Pietrowski D. Smoking decreases endometrial thickness in IVF/ICSI patients. Geburtshilfe Frauenheilkd. 2018;78(1):78-82.
467. Mojtabai R, Crum RM. Cigarette smoking and onset of mood and anxiety disorders. Am J Public Health. 2013;103(9):1656-1665.
468. Feng RM, Hu SY, Zhao FH, et al. Role of active and passive smoking in high-risk human papillomavirus infection and cervical intraepithelial neoplasia grade 2 or worse. J Gynecol Oncol. 2017;28(5):e47.
469. Min KJ, Lee JK, So KA, Kim MK. Association between passive smoking and the risk of cervical intraepithelial neoplasia 1 in Korean women. J Epidemiol. 2018;28(1):48-53.
470. American College of Cardiology. 2018 ACC expert consensus decision pathway on tobacco cessation treatment. J Am Coll Cardiol. 2018;72(25):3332-3365.
471. Tomko RL, Saladin ME, Baker NL, et al. Sex differences in subjective and behavioral responses to stressful and smoking cues presented in the natural environment of smokers. Nicotine Tob Res. 2020;22(1):81-88.
472. U.S. Food and Drug Administration. FDA Announces Comprehensive Regulatory Plan to Shift Trajectory of Tobacco-Related Disease, Death. Available at https://www.fda.gov/news-events/press-announcements/fda-announces-comprehensive-regulatory-plan-shift-trajectory-tobacco-related-disease-death. Last accessed May 3, 2022.
473. Campaign for Tobacco-Free Kids. Raising Cigarette Taxes Reduces Smoking, Especially Among Kids (And the Cigarette Companies Know it). Available at https://www.tobaccofreekids.org/assets/factsheets/0146.pdf. Lat accessed May 3, 2022.
474. U.S. Food and Drug Administration. Statement from FDA Commissioner Scott Gottlieb, M.D., on Actions to Advance our Comprehensive Plan to Reduce Tobacco-Related Disease and Death, Through New Efforts to Improve the Tobacco Product Application Review Process, Including a Newly Proposed Rule. Available at https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-actions-advance-our-comprehensive-plan-reduce-tobacco. Last accessed May 3, 2022.
475. U.S. Food and Drug Administration. The Historic Tobacco Control Act. Available at https://www.fda.gov/tobacco-products/products-guidance-regulations/rules-regulations-and-guidance. Last accessed May 3, 2022.
476. U.S. Food and Drug Administration. Modified Risk Granted Orders. Available at https://www.fda.gov/tobacco-products/advertising-and-promotion/modified-risk-granted-orders. Last accessed May 3, 2022.
477. U.S. Food and Drug Administration. FDA Grants First-Ever Modified Risk Orders to Eight Smokeless Tobacco Products. Available at https://www.fda.gov/news-events/press-announcements/fda-grants-first-ever-modified-risk-orders-eight-smokeless-tobacco-products. Last accessed May 3, 2022.
478. UNDO. Vaping May Increase Your Risk of Cancer. Available at https://www.undo.org/health/vaping-cancer-risk. Last accessed May 4, 2022.
479. Kelley TF. Cigarette Filters and Polonium-210. Available at https://www.industrydocuments.ucsf.edu/tobacco/docs/#id=qjfh0113. Last accessed May 4, 2022.
480. U.S. Food and Drug Administration. FDA Updates and Press Announcements on Nitrosamine in Varenicline (Chantix). Available at https://www.fda.gov/drugs/drug-safety-and-availability/fda-updates-and-press-announcements-nitrosamine-varenicline-chantix. Last accessed March 15, 2022.
481. U.S. Food and Drug Administration. FDA Proposes Rules Prohibiting Menthol Cigarettes and Flavored Cigars to Prevent Youth Initiation, Significantly Reduce Tobacco-Related Disease and Death. Available at https://www.fda.gov/news-events/press-announcements/fda-proposes-rules-prohibiting-menthol-cigarettes-and-flavored-cigars-prevent-youth-initiation. Last accessed May 16, 2022.
482. Brooks M. FDA Clears First Brain Stimulation Device to Help Smokers Quit. Available at https://www.medscape.com/viewarticle/936234. Last accessed May 16, 2022.
1. U.S. Preventive Services Task Force. Primary care interventions for prevention and cessation of tobacco use in children and adolescents: U.S. Preventive Services Task Force recommendation statement. JAMA. 2020;323(16):1590-1598. Available at https://jamanetwork.com/journals/jama/fullarticle/2765009. Last accessed May 11, 2022.
2. U.S. Preventive Services Task Force. Screening for lung cancer: U.S. Preventive Services Task Force recommendation statement. JAMA. 2021;325(10):962-970. Available at https://jamanetwork.com/journals/jama/fullarticle/2777244. Last accessed May 11, 2022.
3. de Ferranti SD, Steinberger J, Ameduri R, et al. Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association. Circulation. 2019;139(13):e603-e634. Available at https://www.ahajournals.org/doi/10.1161/CIR.0000000000000618. Last accessed May 11, 2022.
4. U.S. Preventive Services Task Force. Interventions for tobacco smoking cessation in adults, including pregnant persons: U.S. Preventive Services Task Force recommendation statement. JAMA. 2021;325(3):265-279. Available at https://jamanetwork.com/journals/jama/fullarticle/2775287. Last accessed May 11, 2022.
5. University of Michigan Health System. Tobacco Treatment. Ann Arbor, MI: University of Michigan Health System; 2012. Available at https://www.med.umich.edu/1info/FHP/practiceguides/smoking/smoking.pdf. Last accessed May 11, 2022.
Mention of commercial products does not indicate endorsement.