In the management of patients with mental health disorders, pharmacotherapy remains a vital part of optimal treatment. This course will review the approaches to incorporating pharmacotherapy into the management of the most common mental health disorders in the United States. A variety of potential adverse effects and considerations will also be included.
This course is designed for nurses and pharmacy professionals involved in the care of patients with mental health conditions.
The purpose of this course is to provide members of the interprofessional healthcare team with the information necessary to appropriate prescribe, administer, and dispense psychopharmacotherapy, with the ultimate goal of improving patient care and public health.
Upon completion of this course, you should be able to:
- Outline the history of pharmacology in psychiatry.
- Describe the action and use of typical antipsychotics.
- Compare and contrast the various atypical antipsychotics.
- Evaluate class-wide adverse effects of antipsychotics.
- Identify available antidepressant medication and their use in the treatment of major depression.
- Discuss potential adverse effects and warnings associated with antidepressants.
- Describe medications used in the management of bipolar disorder.
- Assess the role of antidepressants in the management of anxiety disorders.
- Discuss the use of benzodiazepines for the treatment of anxiety.
- Review available pharmacotherapy to incorporate into the treatment of substance use disorders.
Carol Whelan, APRN, has been working in nursing education since 2000. She received her Master's degree in psychiatric/mental health nursing from St. Joseph College in West Hartford, Connecticut, and completed post-graduate nurse practitioner training at Yale University. Ms. Whelan is an Associate Clinical Professor and Lecturer at Yale University and works as an APRN at the Department of Veterans' Affairs in Connecticut, where she also serves as the Vice President of Medical Staff. She has authored many articles, textbook chapters, and books.
Contributing faculty, Carol Whelan, APRN, has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Mary Franks, MSN, APRN, FNP-C
Randall L. Allen, PharmD
The division planners have disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Sarah Campbell
The Director of Development and Academic Affairs has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
The purpose of NetCE is to provide challenging curricula to assist healthcare professionals to raise their levels of expertise while fulfilling their continuing education requirements, thereby improving the quality of healthcare.
Our contributing faculty members have taken care to ensure that the information and recommendations are accurate and compatible with the standards generally accepted at the time of publication. The publisher disclaims any liability, loss or damage incurred as a consequence, directly or indirectly, of the use and application of any of the contents. Participants are cautioned about the potential risk of using limited knowledge when integrating new techniques into practice.
It is the policy of NetCE not to accept commercial support. Furthermore, commercial interests are prohibited from distributing or providing access to this activity to learners.
Supported browsers for Windows include Microsoft Internet Explorer 9.0 and up, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Supported browsers for Macintosh include Safari, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Other operating systems and browsers that include complete implementations of ECMAScript edition 3 and CSS 2.0 may work, but are not supported. Supported browsers must utilize the TLS encryption protocol v1.1 or v1.2 in order to connect to pages that require a secured HTTPS connection. TLS v1.0 is not supported.
The role of implicit biases on healthcare outcomes has become a concern, as there is some evidence that implicit biases contribute to health disparities, professionals' attitudes toward and interactions with patients, quality of care, diagnoses, and treatment decisions. This may produce differences in help-seeking, diagnoses, and ultimately treatments and interventions. Implicit biases may also unwittingly produce professional behaviors, attitudes, and interactions that reduce patients' trust and comfort with their provider, leading to earlier termination of visits and/or reduced adherence and follow-up. Disadvantaged groups are marginalized in the healthcare system and vulnerable on multiple levels; health professionals' implicit biases can further exacerbate these existing disadvantages.
Interventions or strategies designed to reduce implicit bias may be categorized as change-based or control-based. Change-based interventions focus on reducing or changing cognitive associations underlying implicit biases. These interventions might include challenging stereotypes. Conversely, control-based interventions involve reducing the effects of the implicit bias on the individual's behaviors. These strategies include increasing awareness of biased thoughts and responses. The two types of interventions are not mutually exclusive and may be used synergistically.
#95231: Psychopharmacology
Psychiatry and the medications prescribed for psychiatric issues are so intertwined with culture and society that it is impossible to discuss its history without also talking about the societal context and cultural beliefs that surround it. No orthopedic surgeon has ever had to convince a patient that a broken leg was not caused by demons and that the therapy prescribed is medically needed. The same cannot be said for psychiatry. Even in the 21st century, clinicians who deal with the complex medications used to treat psychiatric illness encounter patients who feel shame, guilt, and doubt associated with their diagnoses and the medications used to manage them.
The root of this skepticism can be traced to the French philosopher René Descartes in the 1500s and the famous Cartesian motto "I think therefore I am." According to this widely embraced philosophy, one exists because s/he thinks, not because the heart beats, the muscles work, or the lungs breathe. From this perspective, one is because their brain is healthy.
Given this belief that a disordered mental state calls one's very existence into question, it is no wonder patients are reluctant to seek help, ashamed to report their symptoms, and afraid that they may lose their job or custody of their children—there is an underlying belief that their existence will be questioned if something is wrong with their thoughts, emotions, and the inner core of their being. This is the world in which psychiatric medications are being prescribed. But how did we get here?
In the United States, the first attempt at humane treatment of psychiatric disorders was by the Quakers, who, in 1752, used the Pennsylvania Hospital in Philadelphia to house patients in its basement; however, the environment in the hospital was damaging to patients, some of whom were shackled to walls[1]. Other early institutions for people deemed "mentally disturbed" were opened in the late 1700s in New York. In 1824, the Eastern Lunatic Asylum (now Eastern State Hospital) opened in Lexington, Kentucky. By 1890, every state had at least one publicly supported mental hospital, with populations that were rapidly expanding; by the 1950s, these institutions housed more than 500,000 patients [1]. One of the earliest practitioners of what could be called psychiatry was Dr. Benjamin Rush, who has been called the father of American psychiatry and who wrote the first American textbook on mental diseases (Medical Inquiries and Observations upon Diseases of the Mind) in 1812.
The Association of Medical Superintendents of American Institutions for the Insane was founded in 1844; in 1921, the name changed to the American Psychiatric Association (APA) [2]. In the 1800s, there was no real psychopharmacology or even a true appreciation of the biologic nature of mental illness. However, it is important to acknowledge Rush's decision to refer to "diseases of the mind," rather than more judgmental terminology common to the time, such as spiritual disease or weakness. In fact, Rush hypothesized that psychiatric disorders were caused by irritation of the blood vessels in the brain. This led to treatments such as bleeding and purging and to what could be called the first attempt at the development of a psychoactive medication—the use of mercury as a treatment for mental illness. While it is easy to dismiss such early attempts, this theory of a vascular cause of psychiatric symptoms is consistent with some current knowledge of vascular dementia, and while the theories seem ill-informed today, it is important to note that other popular theories of psychiatric etiology of the time attributed mental illness to demons, witchcraft, or moral turpitude.
During this early period, institutional care of the mentally ill was often a means to persecute women and others whom society wished to confine, which led to what some call the "coercive era" of psychiatric care. One of the most important of these cases was that of Elizabeth Packard in 1860. Elizabeth Packard was married to a clergyman who forcibly placed her in an Illinois asylum. In 1860, Illinois law allowed for involuntary hospitalization of a spouse (generally a wife) by request without any evidence. Ms. Packard was eventually able to obtain release from the hospital, but due to her husband's continuing campaign, she was forced to petition the court and was released in 1863 with a habeas corpus hearing. After gaining her freedom, Ms. Packard began a campaign for the protection of women's rights. As a result of her efforts and the efforts of others at the time, laws were passed requiring a jury trial prior to involuntary hospitalization. It is notable that the Association of Medical Superintendents of American Institutions for the Insane (now the APA) opposed these laws at the time. The damage done by these forcible hospitalizations was huge, and the association of psychiatry with coercion persists to this day. These beliefs influence both the public's acceptance of psychopharmacology as a valid and effective treatment and the acceptance of psychiatric diagnoses as true medical diagnoses, as opposed to an excuse to prescribe medications or force hospitalization.
Another leader in the development of humane custodial care of the psychiatric patients was Dorothea Dix, who, in addition to her other accomplishments, is credited with opening more than 30 state mental hospitals throughout the United States. Dix advocated for humane institutions, particularly for the poor and unhoused, and reformation of mental health treatment.
The landmark court case O'Connor v. Donaldson ruled that the government cannot confine an individual who is not dangerous and is capable of living outside the institution. This ruling was based on the case of Kenneth Donaldson, who in his youth had undergone a course of brief psychiatric care, but had recovered, married, and moved from Florida to Pennsylvania. In 1956, he travelled to Florida to visit his parents, where he made a remark (possibly a joke) that he believed someone in Pennsylvania may have been trying to poison his food. In response, his father hired a lawyer, and at a hearing (where he had no legal representation), Donaldson was civilly committed. He was then placed in a housing unit with dangerous and violent criminals. He received no treatment and, as a Christian Scientist, refused all medications and did not agree that he was experiencing mental illness, which was against the typical routine of admitting illness, accepting treatment, and being released. Donaldson spent more than 15 years in this asylum, despite friends petitioning the administrator for his release and guaranteeing that they would care for him. He eventually won his release by petitioning the courts directly and then proceeded to file suit against the administrator (O'Connor) who had blocked his release, on the constitutional ground of illegal confinement denying him his right to liberty.
The court found that mental illness alone cannot justify custodial confinement against the will of the individual. Further cases (e.g., Lake v. Cameron) introduced the concept of care in the least restrictive setting, and in 1999, the Supreme Court ruled in Olmstead v. L.C. that mental illness is a disability covered under the Americans with Disabilities Act.
At the same time that the courts were acknowledging the rights of persons with mental illness, pharmacologists were making breakthroughs; therapeutic use of chlorpromazine (Thorazine) had been discovered, and the world of psychiatry would never be the same.
The history of psychopharmacology is intertwined with the history of neurology and psychiatry. During the 19th century, psychiatry and neurology were chiefly concerned with the treatment of psychosis and deviant behaviors. The treatment of less severe conditions (e.g., depression, neurosis) was considered outside the province of psychiatry or neurology.
At the beginning of the 20th century, Sigmund Freud published his famous theories on the unconscious roots of what he termed psychoneurosis [3]. He was especially interested in conversion disorders, whereby patients displayed paralysis or other somatic symptoms that could not be explained in a medical context. Freud developed psychoanalysis (talk therapy) and thus began the outpatient treatment of patients with psychiatric concerns.
By the mid-20th century, psychoanalysis became overwhelmingly popular and was employed not only in the treatment of patients with anxiety or neurosis but also in the treatment of patients with more severe psychiatric disorders, such as psychosis, for which talk therapy had little to offer. Some postulate that the development of psychoanalysis stunted the development of psychopharmacology by promoting the notion that talk therapy could effectively treat all mental health issues.
One of the first treatments of psychosis—electroconvulsive therapy or ECT—was developed in the 1930s. Its development arose from the observation that patients with both psychosis and epilepsy seemed to experience improvements in psychotic symptoms following a seizure. While this apparent improvement may have actually been a postictal phase of the seizure disorder, it spurred a search for ways to induce seizures in patients with severe psychiatric disorders. Initial attempts used high doses of stimulant drugs to produce seizures, and the term convulsive therapy was coined. Due to drawbacks evident with the use of stimulant drugs, Italian neurologist Ugo Cerletti experimented with inducing seizures using electrical shocks delivered to the head [4].
Despite its roots in somewhat dubious observations of the effects of electric shocks on livestock, ECT remains in use today as a valid psychiatric treatment. In fact, it was one of the only truly effective psychiatric treatments in the 1930s and 1940s. It was also arguably one of the first treatments to address mental illness as a biologic disorder. Some consider the advent of convulsive therapy as the true beginning of psychopharmacology, as it relied on alteration of brain chemistry to produce improvements in patients.
Building on these advances, psychopharmacology exploded in the 1950s with the development of chlorpromazine, early antidepressants, and early mood stabilizers.
The term psychopharmacology traces its roots to 1920, appearing first in the title of a paper by David Macht describing the use of quinine and antipyretics in tests on neuromuscular coordination. However, most experts date the true birth of psychopharmacology to 1951, when chlorpromazine was first synthesized [5].
Chlorpromazine was developed in the lab of Rhône-Poulenc (an early French pharmaceutical company) for use in general anesthesia to induce calmness. It was not long before French surgeons saw the potential for the use of chlorpromazine in the treatment of psychiatric illness. The first paper on chlorpromazine (titled "A new stabilizer") was published in February 1952 in La Presse Medicale [5].
The first documented clinical psychiatric use of chlorpromazine was on January 19, 1952, when 50 mg intravenous (IV) chlorpromazine was given to a patient, 24 years of age, with severe agitation, psychosis, and possibly mania. The effects were noted as calming but also were transient in nature. After 20 days of treatment with a cumulative total of 900 mg of chlorpromazine along with concomitant barbiturates and ECT, the patient was able to be discharged home [5]. By 1957, chlorpromazine had become internationally recognized, and the American Public Health Association presented Albert Lasker Awards for Medical Research to several scientists and physicians associated with the development of chlorpromazine and introduction of the drug into clinical practice.
Perhaps the greatest contribution of chlorpromazine was not in its clinical effects but rather in its ability to distinguish the biologic basis of schizophrenia. During the course of the 1950s, studies of chlorpromazine use led to an understanding of synaptic transmission and development of the theory that synaptic transmission was not a merely electrical event but rather a chemically mediated event. By the end of the 1950s, six neurotransmitters had been identified in the central nervous system, including dopamine, norepinephrine, and serotonin [5].
The success of chlorpromazine led to the development of more antipsychotics, including thioridazine (Mellaril), haloperidol (Haldol, decanoate), trifluoperazine (Stelazine), and fluphenazine (Prolixin). However, all of these early antipsychotics were noted to have neurologic side effects, most notably extrapyramidal symptoms with parkinsonian features.
Some consider the 1950s to be a "golden age" of psychopharmacology. Beginning in 1958, tricyclic compounds based on the structure of imipramine were synthesized; these are referred to as second-generation (atypical) antipsychotics. One of these compounds was clozapine (Clozaril, Versacloz), an antipsychotic still in use today. Interestingly, clozapine did not initially attract much attention. Small trials showed mixed results, and unlike chlorpromazine, there was no consensus as to its effectiveness. The lack of extrapyramidal side effects led many researchers to erroneously believe that it was not a true antipsychotic, as the dopamine blockade theory of schizophrenia was still widely held.
The development of medications to treat psychiatric illnesses has greatly informed our understanding of the biologic nature of these illnesses—most notably the link between antipsychotics and schizophrenia. This translational research also led to the understanding of neurotransmitters and further informed the development of large classes of psychoactive medications.
The dopamine receptor D2 is a common target of the antipsychotics. Assessment of medications' ability to target receptors was made possible by the introduction of the spectrophotofluorimeter in 1955. This instrument allows for analysis of chemicals in the brain, including monoamines, which allowed for the further advancement of the field of psychopharmacology [5].
With their introduction in the 1950s, monoamine oxidase inhibitors (MAOIs) became the first class of medications used to treat depression. The incorporation of these agents into clinical practice further moved psychiatry away from inpatient care in institutions toward effective outpatient care.
Another historical breakthrough in psychopharmacology was the discovery of the use of lithium for the treatment of mania. While there is some evidence of usage in the 1800s, the seminal research reintroducing lithium for the treatment of mania was published in 1949 by John Cade [6]. Despite its ready availability and widespread use worldwide, lithium was not approved by the U.S. Food and Drug Administration (FDA) until 1970; the United States was the 50th country to approve the use of lithium for the management of mania [6].
The discovery of medications effective in the treatment of affective disorders induced a shift away from the talk therapy-only approach originally developed by Freud. Further, the ability of lithium to improve symptoms of depression supported the burgeoning biologic theory of mental illness. This change in understanding of mental illness had extensive and widespread influence on the practice of psychiatry. It extended beyond schizophrenia and bipolar disorder and helped transform etiologic theories of diseases such as autism from a focus on environmental influence (e.g., poor mothering in the case of autism) to a biologic understanding of psychiatric disorders prevalent today.
In the later part of the 20th century, psychopharmaceutical advancements continued at record pace. The development of benzodiazepines, lithium, barbiturates, and MAOIs led to an era of deinstitutionalization and the mainstream diagnosis and management of psychiatric disorders without a reliance on talk therapy. This seismic change was not without controversy, and proponents of talk therapy and psychoanalysis resisted the use of psychopharmacology to treat mental illness.
This philosophical conflict was clearly evident in the revision process for the APA's Diagnostic and Statistical Manual of Mental Disorders (DSM). In 1980, the DSM was extensively revised (creating the DSM-III), and psychoanalytic language was abandoned. In contrast to the DSM-II, the DSM-III focused on symptom-based descriptions of mental disorders for diagnostic criteria.
Psychiatry continued to evolve to accept the biologic basis of mental illness and embrace the use of pharmacologic treatment. The introduction of selective serotonin reuptake inhibitors (SSRIs), which were both well-tolerated and had better safety profiles than previous medications, revolutionized the treatment of depression. The first SSRI, fluoxetine (Prozac), was approved by the FDA in 1987. While there has been controversy regarding the possible overdiagnosing of depression or overprescribing of antidepressants, no one disputes the enormous impact fluoxetine has had on psychopharmacology [3].
At virtually the same time as the introduction of fluoxetine, a group of second-generation (atypical) antipsychotics were being approved in the United States. These medications, including risperidone (Perseris, Risperdal) and olanzapine (Zyprexa), challenged the traditional belief that only the D2 receptor/dopamine was involved in psychosis. This renewed interest in the forgotten medication clozapine and novel treatment approaches for schizophrenia.
While clozapine had been originally introduced in the 1950s and approved by the FDA in 1971, small studies had been unable to show efficacy. Use of the drug effectively ended in 1975, when the Finnish National Board of Health reported 18 individuals prescribed clozapine who developed severe blood disorders (most commonly agranulocytosis, a severe decrease or an absence of white blood cells increasing the risk of potentially fatal infection); 9 of these patients died [7]. Research since that time indicates that agranulocytosis occurs in about 1% of patients taking clozapine and neutropenia is seen in about 3% [8].
These early roadblocks to the use of clozapine were overcome, first by a system of monitoring white blood cell counts and secondly by longer studies that showed that the effects of clozapine continue to increase after four weeks of therapy, unlike chlorpromazine, which had a peak effect after two to three weeks [7]. As a result of these findings, clozapine was approved for the treatment of psychosis in the United States in 1990 [7]. The price of this drug when it was first approved, approximately $9,000 per patient per year for the medication and monitoring, was initially controversial, but it was more cost-effective than inpatient care or institutionalization [3,7].
The risk for leukopenia remains a barrier to wider use of clozapine; however, it has become a mainstay for treatment-resistant schizophrenia and is also acknowledged to reduce the risk of suicide in patients with schizophrenia. It is also used off-label in the management of treatment-resistant bipolar disorder and dementia- or Parkinson-related psychosis or agitation. With the availability of a generic version, the price of clozapine is now less than many other newer antipsychotics. Knowledge that the risk of agranulocytosis is highest in the first three months has also led to a reduction in the frequency of long-term monitoring.
Substance use disorder is a significant issue in the United States, affecting approximately 48.5 million individuals annually, or 17.1% of the population [9]. Historically, addiction was considered a moral failing—a symptom of criminality or weak character. A variety of non-effective treatments were used until the 1900s, ranging from cocaine-based therapy, hydrotherapy, and institutionalization. In the 1930s, 12-step and self-help models were established for alcohol dependence treatment. In 1949, disulfiram (Antabuse) became the first drug approved to treat alcoholism [10]. The drug works by increasing the concentration of acetaldehyde, a toxic byproduct that occurs when alcohol is broken down in the body. Excess amounts of this byproduct (as when alcohol is consumed along with the medication) cause unpleasant symptoms, such as nausea and flushing of the skin. The anticipation of these effects can assist with cessation and abstinence. For more than 40 years, disulfiram was the only medication approved for the treatment of alcohol use disorder [10]. Since then, two additional medications—naltrexone (ReVia, Vivitrol) and acamprosate (Campral)—have been approved. The conceptualization of addiction and substance use disorder has also evolved and is now recognized to be a primary, chronic disease of brain reward, motivation, memory, and related circuitry. Dysfunction in these circuits leads to characteristic biologic, psychological, social, and spiritual manifestations [11]. As a primarily biologic illness, addiction is now recognized as a valid target for pharmacotherapy.
In the early 21st century, the United States experienced an unprecedented increase in opioid prescribing, use disorder, and fatal overdose [12]. In addition to the highly beneficial therapeutic effects, the toxic side effects and addictive potential of opioids have been known for centuries. These undesired effects have prompted a search for a potent synthetic opioid analgesic free of addictive potential and other complications. However, all synthetic opioids introduced into medical use share the same abuse liabilities of the classical opioids. The search for new opioid therapeutics has resulted in the synthesis of opioid antagonists and compounds with mixed agonist-antagonist properties, such as buprenorphine, which has expanded therapeutic options and provided the basis of expanded knowledge of opioid mechanisms [13].
Nonmedical use of prescription opioids was reported in literature as early as 1880. A report in 1928 documented that injection of opioids contributed to the development of nonmedical use and misuses of prescription opioids. Before 1930, the prevalence of nonmedical opioid injecting in the United States was low. But by the mid-1940s, more than one-half the admissions to the National Institute of Mental Health's Lexington Hospital were for the misuse of prescription opioids [14]. As of 2018, there were an estimated 2.1 million individuals with opioid use disorder in the United States [15]. In addition, more than 81,000 drug overdose deaths occurred in the United States in the 12 months ending in May 2020, the highest number of overdose deaths ever recorded in a 12-month period, and the rise was mainly attributed to synthetic opioids [16].
While methadone has long been in use in specialized clinics with strict licensing, the development of alternative treatments for opioid use disorder, including buprenorphine/naloxone (Suboxone), has revolutionized the approach to managing and treating this disorder.
The American Psychiatric Association recommends that patients with schizophrenia be treated with an antipsychotic medication and monitored for effectiveness and side effects. This guideline statement should be implemented in the context of a person-centered treatment plan that includes evidence-based nonpharmacological and pharmacological treatments for schizophrenia.
(https://psychiatryonline.org/doi/full/10.1176/appi.books.9780890424841.Schizophrenia02 Last Accessed: September 27, 2024)Strength of Recommendation/Level of Evidence: IA (High confidence that the evidence reflects the true effect)
With the development of clozapine, antipsychotic medications have been divided into typical antipsychotics (also referred to as first-generation antipsychotics) and atypical antipsychotics (also referred to as second-generation antipsychotics). In addition, a group of novel atypical antipsychotics have been developed and generally act on serotonin as well as dopamine receptors.
The choice of antipsychotic should be guided by the side effect profile, available route of administration (e.g., liquid forms, oral disintegrating tablets), and the patient's medical history, current medications, and preference. Many patients will be unwilling to switch from a drug that has been effective, even if a different drug may yield better results.
Typical antipsychotics (Table 1) have a primary site of action at the D2 receptor and are potent D2 receptor blockers. They also have noradrenergic, cholinergic, and histaminergic blocking action. These agents can also be described as high, intermediate, or low potency. High-potency antipsychotics are prescribed in lower doses, with the most widely prescribed of these being haloperidol (Haldol). Haloperidol is available in oral, intramuscular (IM), and long-acting IM decanoate formulations. Low-potency antipsychotics are prescribed in higher doses; the most widely used of these is chlorpromazine [17,18].
TYPICAL ANTIPSYCHOTIC MEDICATIONS
| Drug | Dose Range | Typical Starting Dose | Usual Maintenance Dosea | Route(s) | Indication(s) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chlorpromazine | 25–800 mg/day | 25–50 mg/day | 100–400 mg daily or BID | IM, IV, PO | Schizophrenia, bipolar disorder, intractable hiccups, agitation/aggression (severe, acute) associated with psychiatric disorders | ||||||
| Droperidol | 0.625–10 mg/day | 2.5–10 mg/day | 10 mg/day | IM, IV | Postoperative nausea/vomiting, acute undifferentiated agitation (off-label) | ||||||
| Flupentixol |
|
|
| Oral, IM (depot) | Schizophrenia | ||||||
| Fluphenazine | 2.5–40 mg/day | 2.5–10 mg every 6 to 8 hours |
| PO, IM, decanoate | Psychotic disorders | ||||||
| Haloperidol (Haldol) | 0.5–30 mg/day | Oral: 0.5–5 mg BID | 5–15 mg BID | PO, IM, IV, decanoate | Bipolar disorder, hyperactive delirium, schizophrenia, Tourette-associated tics, acute/severe agitation (off-label) | ||||||
| Loxapine (Adasuve) | 5–125 mg BID | 10–25 mg BID | 60–100 mg BID | PO, inhalation | Schizophrenia, acute agitation | ||||||
| Methotrimeprazine | 6–200 mg/day |
|
| PO, IM | Anxiety/tension disorders, insomnia, nausea/vomiting, pain, psychotic disorders | ||||||
| Molindone | 5–75 mg TID | 5–15 mg BID | 10–25 mg TID | PO | Schizophrenia | ||||||
| Periciazine | 5–40 mg/day | 5–20 mg in the morning, followed by 10–40 mg in the evening | Titrate to lowest effective dose | PO | Psychosis | ||||||
| Perphenazine | 2–24 mg BID | 2–4 mg BID | 8–24 mg BID | PO | Schizophrenia, nausea/vomiting | ||||||
| Pimozide | 0.5–10 mg/day | 1–2 mg/day in divided doses | Lowest effective dose (maximum: 10 mg) | PO | Tourette syndrome, delusional infestation (off-label) | ||||||
| Prochlorperazine (Compro) | 2.5–25 mg/day |
| Maximum: 40 mg/day | PO, IM, IV, rectal | Acute nausea and vomiting | ||||||
| Thioridazine | 50–800 mg/day | 50–100 mg TID | 200–800 mg BID or QID | PO | Schizophrenia | ||||||
| Thiothixene | 2–30 mg BID | 2–5 mg BID | 10–15 mg BID | PO | Schizophrenia | ||||||
| Trifluoperazine | 2–40 mg/day | 1–2 mg BID | 15–20 mg/day | PO | Schizophrenia | ||||||
| Zuclopenthixol | 10–400 mg/day |
|
| PO, IM | Schizophrenia, psychoses (acute and long-term) | ||||||
| |||||||||||
The adverse and side effect profiles of typical antipsychotics are generally poorer than the atypical antipsychotics. The prevention, detection, and treatment of extrapyramidal symptoms, most notably tardive dyskinesia, is the main challenge when prescribing typical antipsychotics.
Neurologic Effects
Patients who are prescribed typical antipsychotics should be monitored for neurologic side effects using the standardized Abnormal Involuntary Movement Scale (AIMS) (Figure 1). The AIMS consists of 12 items and can usually be completed within 10 minutes. It was developed specifically to detect and record the occurrence of tardive dyskinesia in any patient taking neuroleptic medication. Tardive dyskinesia is a syndrome characterized by abnormal involuntary movements of the patient's face, mouth, trunk, or limbs, and it affects 20% to 30% of patients who have been treated for months or years with neuroleptic medications. Patients who are older, are heavy smokers, or have diabetes are at increased risk of developing tardive dyskinesia. For most patients, this side effect develops three months after the initiation of neuroleptic therapy; in elderly patients, however, tardive dyskinesia can develop after as little as one month [20].
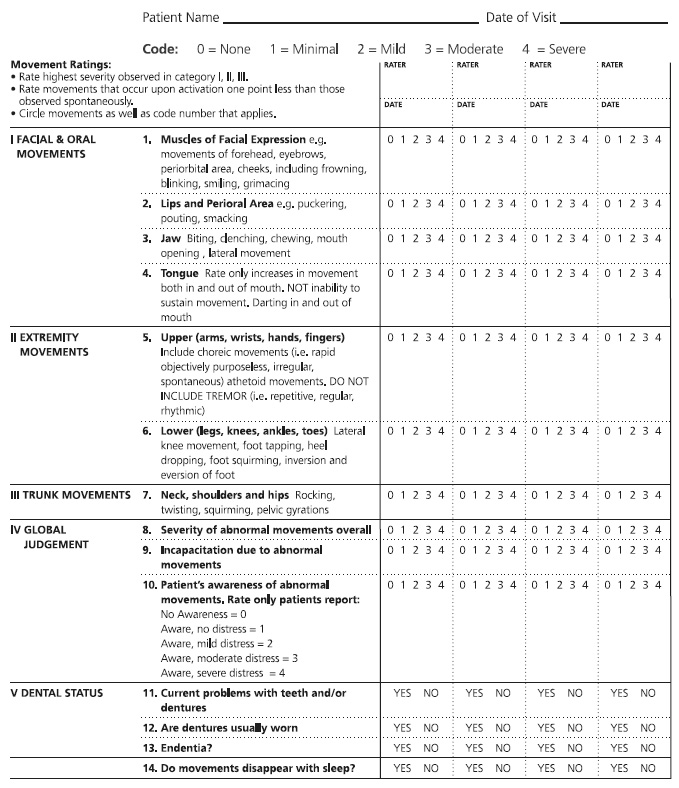
Regular use of the AIMS allows the severity of symptoms to be followed over time. Initial AIMS testing should be done prior to the initiation of treatment and every three to six months during therapy. Either before or after completing the examination procedure, the patient should be observed unobtrusively at rest (e.g., in the waiting area). The chair used in the examination should be firm and without arms. The patient should remove his or her shoes and socks and any gum, candy, or food from his/her mouth prior to the examination. In addition to the examination and observation, the patient should be asked about:
Dental/oral health (e.g., use of dentures, problems with teeth)
Unintended movements in the extremities, mouth, or face and whether these movements are a bother or interfere with activities
Patients should be instructed to sit in a chair with their feet flat on the ground; a general observation of the patient's body may be made at this point. Patients should also be asked to drape their arms over their legs, with their hands hanging over their knees. Several additional tasks should be done, including:
Opening the mouth to observe whether the tongue is moving or at rest
Sticking the tongue out to observe for any abnormal movements
Tapping the thumb with each finger as rapidly as possible to observe the flexibility of fingers as well as any other body movements that occur while the patient is concentrating on this manual task
Flexing and extending the arms and wrists one at a time to monitor for rigidity and cogwheeling
Walking around the room to observe for any parkinsonian-type movements
All findings during the examination should be recorded. It is important to note that repetitive, regular, rhythmic tremors are not recorded on the AIMS, as the tool is specific to tardive dyskinesia. However, these tremors may in fact be an adverse effect of neuroleptic treatment. If present, these movements should be monitored and considered when making treatment and pharmacotherapy decisions.
Metabolic Effects
Meta-analyses have shown that all antipsychotics are associated with an increased risk for weight gain, with the greatest gain seen with atypical agents (clozapine and olanzapine) and the least with typical agents (haloperidol). Numerous studies have concluded that H1 receptor antagonism action is most correlated with weight gain, although other receptor sites (including 5HT receptors) were also significantly associated with weight gain [21].
A reasonable approach to monitoring metabolic side effects involves measuring vital signs and weight prior to prescribing an antipsychotic agent and at every follow-up visit. Baseline blood work including a lipid profile, hemoglobin A1C, and a basic metabolic panel is also advised. Variances from baseline values and the presence or absence of weight gain can help guide the frequency of monitoring. However, obtaining laboratory studies at least yearly should be considered.
Cardiovascular Effects
Monitoring for QTc prolongation should be a consideration for all patients prescribed an antipsychotic. Obtaining a baseline electrocardiogram (EKG) prior to prescribing is good practice. Values greater than 500 msec should prompt referral to cardiology and a consideration to avoid medications that can prolong the QTc interval. Values of 450–500 msec require close follow-up EKG monitoring both after initiation of treatment and during any dosage adjustments.
Antipsychotics remain a consistently popular option for the management of psychosis and schizophrenia. Since the reintroduction of clozapine in the 1990s, there has been a steady increase in the proportion of atypical antipsychotics prescribed compared with typical antipsychotics. By 2014, atypical antipsychotics accounted for almost 80% of total antipsychotics prescribed [22]. This has been partially attributed to treatment failure with typical antipsychotics (e.g., up to 30% of patients with schizophrenia fail to respond to typical antipsychotics), a desire to avoid extrapyramidal symptoms, and expansion of FDA-approved indications for atypical agents [17]. It should be noted that some of the lesser-used, low-potency typical antipsychotics may also avoid extrapyramidal effects.
As of 2024, there were 14 atypical antipsychotics approved for use in the United States (Table 2). Like the typical agents, these drugs are used primarily for the treatment of psychoses, although several are adjunctive treatments for major depressive disorder. Novel antipsychotics with various mechanisms of actions continue to be studied and approved; clinicians should consult the latest prescribing literature when selecting a medication for their patient.
ATYPICAL ANTIPSYCHOTIC MEDICATIONS
| Drug | Dose Range | Typical Starting Dose | Usual Maintenance Dosea | Route(s) | Indication(s) | ||
|---|---|---|---|---|---|---|---|
| Aripiprazole (Abilify) | 2–30 mg/day | 2.5–5 mg/day | 10–20 mg/day | PO, decanoate | Bipolar disorder, treatment-resistant depression (adjunctive), schizophrenia | ||
| Asenapine (Saphris, Secuado) | 5–10 mg BID | 5 mg BID | 10 mg BID | SL, transdermal patch | Bipolar disorder, schizophrenia | ||
| Brexpiprazole (Rexulti) | 2–4 mg/day | 1 mg/day | 4 mg/day (with titration) | PO | Schizophrenia, treatment-resistant depression (adjunctive) | ||
| Cariprazine (Vraylar) | 1.5–6 mg/day | 1.5–6 mg/day | 3–6 mg/day (with titration) | PO | Bipolar disorder, schizophrenia | ||
| Clozapine (Clozaril, Versacloz) | 25–800 mg/day | 25–800 mg/day | 250–400 mg hourly or BID (Strict titration guidelines, must be in REMS) | PO (including oral disintegrating tablet) | Schizophrenia, suicidal behavior in schizophrenia or schizoaffective disorder | ||
| Iloperidone (Fanapt) | 1–12 mg/day | 1–2 mg BID | 12 mg/day (lower doses in patients with hepatic dysfunction) | PO | Schizophrenia | ||
| Lumateperone (Caplyta) | 42 mg/day | 42 mg/day | 42 mg/day | PO | Schizophrenia | ||
| Lurasidone (Latuda) | 20–80 mg/day | 20 mg/day | 80 mg/day | PO | Bipolar major depression, schizophrenia | ||
| Olanzapine (Zyprexa) | 2.5–20 mg/day | 2.5–5 mg/day | 15–20 mg/day | PO, IM, IV | Bipolar disorder, agitation/aggression associated with psychiatric disorders, schizophrenia | ||
| Paliperidone (Invega) | 3–12 mg/day | 3 mg/day | 6 mg/day | PO, IM decanoate | Schizophrenia, schizoaffective disorder | ||
| Pimavanserin (Nuplazid) | 34 mg/day | 34 mg/day | 34 mg/day | PO | Parkinson-associated psychosis | ||
| Quetiapine (Seroquel) | 25–800 mg/day | 50 mg/day | 200–400 mg/day | PO, extended-release | Bipolar disorder, schizophrenia | ||
| Risperidone (Perseris, Risperdal) | 0.5–6 mg/day | 1–2 mg/day | 4 mg/day | PO, IM decanoate | Bipolar disorder, schizophrenia | ||
| Ziprasidone (Geodon) | 20–80 mg/day | 40 mg BID | 80 mg BID | PO, short-acting IM | Bipolar disorder (adjunctive), schizophrenia | ||
| |||||||
When choosing an atypical antipsychotic, it is important that the clinician fully understand the mechanism of action of the drug being considered, as they have a wide variety of actions. There are also significant pharmacokinetic differences.
Aripiprazole is a partial dopamine agonist (not a blocker). Some research indicates that this medication may have a lower risk of tardive dyskinesia and metabolic syndrome. The initial effects of the oral preparation may be observed within days of treatment of bipolar disorder or acute mania and within one to two weeks in those with major depressive disorder or schizophrenia [19]. In all patients, continued improvement in efficacy is noted over the next several weeks. For bipolar/acute mania, maximal efficacy is seen in one to two weeks [19]. However, those with schizophrenia may see improvements for 4 to 6 weeks, and those with major depressive disorder may see maximal effects after 6 to 12 weeks [19].
Aripiprazole is metabolized hepatically and is primarily excreted in the feces (55%) and urine (25%). Peak plasma levels are noted in three to five hours following oral tablet administration, though high-fat meals can delay this time by hours. Following multiple IM doses of the extended-release formulation, peak plasma levels are noted in four days after deltoid administration or after five to seven days following gluteal administration [19].
The only absolute contraindication to the use of aripiprazole is hypersensitivity (including anaphylaxis) to the drug or any components of the formulation. The drug is associated with orthostatic hypotension and should be used with caution in patients predisposed to this effect or who are unable to tolerate transient hypotensive episodes [19]. Other possible adverse effects include increased serum glucose, weight gain, constipation, tremor, nausea/vomiting, agitation, anxiety, sedation/drowsiness, extrapyramidal reactions, headache, and insomnia [19].
Asenapine is a dibenzo-oxepino pyrrole atypical antipsychotic with mixed serotonin-dopamine antagonist activity. It is unique as the only atypical antipsychotic available in a sublingual formulation. When administered sublingually for the management of agitation, the onset of effects is seen within 15 minutes. In bipolar disorder/acute mania, initial effects are present within days, with continued improvements over one to two weeks [19]. In patients being treated for schizophrenia, the initial effects are observed within one to two weeks, and maximal effects are noted in four to six weeks.
Asenapine is metabolized by the liver and excreted primarily in the urine and feces [19]. Peak plasma levels are attained within 0.5 to 1.5 hours for the sublingual formulation and within 12 to 24 hours for the transdermal patch.
Severe hepatic impairment (Child-Pugh class C) and hypersensitivity are considered absolute contraindications to asenapine. Possible adverse effects include drowsiness/fatigue, insomnia, extrapyramidal reactions, headache, weight gain, increased serum triglycerides and serum glucose, and oral hypoesthesia [19].
Brexpiprazole exhibits partial agonist activity for 5-HT1A and D2 receptors and antagonist activity for 5-HT2A receptors. As a partial dopamine agonist, it is associated with a decreased risk for extrapyramidal reactions (including tardive dyskinesia) and metabolic syndrome. The onset of action for the oral formulation is typically within 1 to 2 weeks, with increased efficacy over the following weeks (4 to 6 weeks in those with schizophrenia and 6 to 12 weeks in those with major depressive disorder) [19].
Brexpiprazole undergoes hepatic metabolism, primarily by CYP3A4 and CYP2D6. It is excreted in the feces and urine. Brexpiprazole reaches peak plasma levels within four hours [19].
The only absolute contraindication to the use of brexpiprazole is hypersensitivity reactions. However, it should be used with caution in patients with renal impairment. The most common adverse effects of brexpiprazole are increased serum triglycerides (typically <500 mg/dL), weight gain, and akathisia (inability to remain still) [19].
Cariprazine acts as a partial dopamine (D2) and serotonin (5-HT1A) agonist, with antagonist activity at serotonin 5-HT2A receptors. Onset of initial effects varies from days (with bipolar disorder/acute mania) to up to two weeks (with schizophrenia) [19]. As with the other atypical antipsychotics, effects increase over time to peak efficacy experienced in up to 12 weeks.
Cariprazine is extensively metabolized by CYP3A4 and, to a lesser extent, by CYP2D6 to active metabolites [19]. It is excreted through several routes, but most prominently through the urine. Peak plasma levels occur within three to six hours. Due to the long half-life of cariprazine and its active metabolites, changes in dose will not be fully reflected in plasma for several weeks.
There are no absolute contraindications to the use of cariprazine aside from hypersensitivity. Common adverse effects include extrapyramidal reactions, parkinsonian-like syndrome, akathisia, headache, nausea, and insomnia [19].
Clozapine has demonstrated efficacy in reducing the risk of suicide in patients with schizophrenia and known efficacy in treatment-resistant schizophrenia. It is believed to act through antagonism of the D2 and serotonin type 2A (5-HT2A) receptors [19]. The onset of action for the oral formulation is observed from within days up to two weeks, with increased effects for the subsequent weeks. For patients with treatment-resistant schizophrenia, longer trials of 8 to 12 weeks are recommended [19].
Metabolism of clozapine is extensively hepatic, with urinary and fecal excretion. Time to peak plasma levels is 2.2 to 2.5 hours, depending on the route of administration [19].
Serious hypersensitivity to clozapine or any component of the formulation is considered an absolute contraindication to its use [19]. Though not included in the U.S. labeling, Canadian labels include myeloproliferative disorders, impaired bone marrow function, active hepatic disease, severe renal impairment, paralytic ileus, uncontrolled epilepsy, severe nervous system dysfunction, and severe cardiovascular disease as additional contraindications. Common adverse effects are hypertension, hypotension, tachycardia, increased serum glucose levels, dyslipidemia, weight gain, constipation, decreased gastrointestinal motility, dyspepsia, nausea/vomiting, sialorrhea, dizziness, drowsiness/sedation, insomnia, vertigo, and fever [19].
The Clozapine REMS Program
Clozapine is associated with severe neutropenia (absolute neutrophil count [ANC] less than 500/mcL). The requirements to prescribe, dispense, and receive clozapine are incorporated into a single shared program called the Clozapine Risk Evaluation and Mitigation Strategy (REMS) Program. A REMS is a strategy to manage known or potential risks associated with a drug or group of drugs and is required by the FDA for clozapine to ensure that the benefits of the drug outweigh the risk of severe neutropenia.
The Clozapine REMS Program provides a centralized point of access for prescribers and pharmacies to certify before prescribing or dispensing clozapine and to enroll and manage patients on clozapine treatment [23]. The monitoring requirements are established for the general population (most patients being prescribed clozapine) and for patients with benign ethnic neutropenia (BEN). BEN is a condition observed in certain ethnic groups whose average ANCs are lower than "standard" laboratory ranges for neutrophils. It is most commonly observed in individuals of African descent (approximate prevalence of 25% to 50%), some Middle Eastern ethnic groups, and in other non-White ethnic groups with darker skin; it is also more common in men [23]. At initiation of therapy, ANC monitoring should be conducted weekly for six months. If ANC levels remain ≥1,500/mcL in the general population or ≥1,000/mcL in patients with BEN, monitoring frequency may be reduced to every two weeks from 6 to 12 months, then monthly after 12 months. If neutropenia develops, monitoring may be more frequent, from three times weekly to daily depending on the severity of the neutropenia [23]. Prescribers are required to submit patients' ANC levels to the Clozapine REMS Program for every prescription of clozapine according to the patient's monitoring frequency.
Iloperidone exhibits mixed dopamine (D2)/serotonin (5-HT2) antagonist activity. The agent's low affinity for histamine H1 receptors may decrease the risk for weight gain and somnolence, and its affinity for norepinephrine α1/α2C may improve cognitive function but increase the risk for orthostasis. With oral administration, the onset of antipsychotic action may be observed within one to two weeks of treatment, with four to six weeks to peak effect [19].
Hepatic metabolism of iloperidone results in creation of the active metabolites P88 and P95. These metabolites are excreted primarily in the urine and, less extensively, in the feces. Peak plasma levels are achieved in two to four hours [19].
Contraindications are limited to hypersensitivity reactions. The most common adverse effects are tachycardia, dizziness, drowsiness, increased serum prolactin, and weight gain [19].
Lurasidone has documented mixed serotonin-dopamine antagonist activity, with high affinity for D2, 5-HT2A, and 5-HT7 receptors. Improvements in psychosis or bipolar disorder/depressive episode are noted within one to two weeks of treatment, although efficacy will improve for up to six weeks [19]. The medication should be given with food to improve absorption.
Hepatic metabolism is achieved primarily via CYP3A4 and results in two main active metabolites and two main nonactive metabolites [19]. Excretion is mainly in feces. Peak serum concentration occurs within one to three hours, with steady state concentrations achieved within seven days.
Lurasidone is contraindicated with hypersensitivity reactions or concomitant use with strong CYP3A4 inhibitors (e.g., ketoconazole, clarithromycin, ritonavir, voriconazole, mibefradil) and inducers (e.g., rifampin, avasimibe, St. John's wort, phenytoin, carbamazepine) [19]. It should be used with caution and at the lowest possible dose in patients with hepatic impairment. Other common adverse effects include dyslipidemia, increased serum glucose, nausea, extrapyramidal reaction, drowsiness, akathisia, parkinsonian-like syndrome, and insomnia.
Olanzapine displays potent antagonism of serotonin, dopamine, histamine, and alpha1-adrenergic receptors [19]. Its antipsychotic action is believed to be related to its effect on dopamine and serotonin. For acute agitation, effects are seen within 15 minutes with IM injection or within 5 to 10 minutes with IV administration. When administered orally for the management of bipolar disorder/acute mania or schizophrenia, initial effects are seen within days to two weeks; improvements may be noted for up to six weeks [19]. This agent is available in a long-acting injection formulation, and details about managing patients prescribed this route will be discussed in detail later in this course.
Olanzapine is metabolized via direct glucuronidation and cytochrome P450-mediated oxidation; 40% is removed via first-pass metabolism [19]. It is excreted in the urine (57%) and feces (30%). Maximum plasma concentrations after IM administration are five times higher than maximum plasma concentrations produced by an oral dose [19]. Clearance is increased in cigarette smokers and decreased in female patients. Following short-acting injection, peak plasma concentrations occur in 15 to 45 minutes; with extended-release injection, peak levels are noted in about one week. Following oral administration, peak serum concentrations are noted within six hours [19].
While there are no published contraindications to olanzapine, hypersensitivity is a likely barrier to use. This drug is associated with dose-related increases in prolactin levels, with associated menstrual-, sexual-, and breast-related effects [19]. Adverse reactions seen most commonly include dyslipidemia, increased serum glucose, weight gain, increased appetite, xerostomia, decreased serum bilirubin, akathisia, dizziness, drowsiness/fatigue, extrapyramidal reactions, headache, insomnia, parkinsonism, and asthenia [19].
As with the other atypical antipsychotics, the antipsychotic effects of quetiapine are believed to be the result of its antagonism of dopamine (D2) and serotonin (5-HT2) receptors [19]. It also has antagonistic action against 5-HT1A, D1, histamine (H1), and adrenergic alpha1- and alpha2-receptors, some of which may result in adverse effects. After oral administration, initial effects are seen within days (for bipolar disorder/acute mania or bipolar disorder/depressive episode) or up to two weeks (for schizophrenia). Continued improvement in symptoms should be expected for 1 to 12 weeks [19]. If the extended-release formulation is used in the management of generalized anxiety disorder, the initial effects are observed within four to seven days, with continued improvement for eight weeks.
Quetiapine metabolism is primarily hepatic, with mainly urinary elimination. The time to peak plasma level is 1.5 hours for the immediate-release version and 6 hours for the extended-release formulation [19].
There are no absolute contraindications to the use of quetiapine, aside from hypersensitivity. Drowsiness is a common effect and may be severe enough to result in impairment, inability to safely carry out activities of daily living (increased fall risk), and nonadherence [19]. Other common adverse effects include hypertension, orthostatic hypotension (particularly among elderly patients), tachycardia, dyslipidemias, weight gain, increased appetite, xerostomia, agitation, dizziness, extrapyramidal reaction, headache, and withdrawal syndrome.
Risperidone's high 5-HT2 and dopamine-D2 receptor antagonist activity is responsible for its antipsychotic action. However, alpha1, alpha2 adrenergic, and histaminergic receptors are also antagonized with high affinity. Onset of action is within days to 2 weeks, though peak effect may be experienced after up to 12 weeks [19]. An orally disintegrating tablet may be used to manage acute agitation, with 70 minutes mean time to calm.
Risperidone undergoes extensive hepatic metabolism to create the active metabolite 9-hydroxyrisperidone [19]. It is primarily excreted in the urine. Time to peak plasma level is about 1 hour with oral administration (but it is 3 to 17 hours for the active metabolite). When administered subcutaneously, the first peak is in 4 to 6 hours; the second peak is in 10 to 14 days.
Hypersensitivity to risperidone, paliperidone, or any component is an absolute contraindication. Following each subcutaneous injection, a lump may develop and persist for several weeks. This is a self-limiting effect, and the injection site should not be rubbed or massaged [19]. Other common adverse effects include hyperprolactinemia, weight gain, constipation, nausea/vomiting, upper abdominal pain, akathisia, anxiety, dizziness, drowsiness/fatigue, extrapyramidal reaction, headache, insomnia, parkinsonism, and tremor.
A subset of antipsychotics (both typical and atypical) are available as long-acting injectable forms (Table 3). When prescribing a long-acting injectable antipsychotic, clinicians first initiate treatment with the oral form for a long enough period to demonstrate tolerance. The oral formulation should be continued until the long-acting therapy has been established. Both fluphenazine and haloperidol use sesame oil in their suspension, which may cause allergic reactions in patients sensitive to sesame.
LONG-ACTING INJECTABLE ANTIPSYCHOTICS
| Drug | Usual Dosing | Onset | Oral Overlap | Comments | ||
|---|---|---|---|---|---|---|
| Fluphenazine decanoate | 25 mg every 3 weeks | 24 hours | 24 hours | — | ||
| Haloperidol decanoate | 50–200 mg every 4 weeks | One week | 7 to 14 days | — | ||
| Paliperidone palmitate | 117–156 mg monthly OR 273–819 mg every 3 months OR 1,092–1,560 mg every 6 months | — | None |
| ||
| Risperidone (Risperdal Consta) | 12.5–25 mg every 14 days | 7 days | 3 weeks | — | ||
| Olanzapine (Zyprexa Relprevv) | 300 mg every 4 weeks | 7 days | None | Post-injection syndrome limits usage. | ||
| Aripiprazole | 300–400 mg every 30 days | 7 days | 7 days | High cost may limit access. |
There are no randomized controlled trials showing superiority of decanoate injections over other formulations; however, some small studies have shown at least similar efficacy of long-acting injected and oral haloperidol [24]. Plasma serum concentrations with steady-state decanoate yield lower plasma drug concentrations than with oral administration. This suggests that decanoate forms are at least as effective as oral agents and that lower doses may be effectively used with this formulation [24].
The efficacy of long-acting injectable antipsychotics other than haloperidol is not clear [25]. Studies have found varying results for different antipsychotics, both in terms of efficacy and side effect profile [24,25]. The ultimate decision to select a long-acting injectable antipsychotic is complicated and typically driven by a need to improve compliance.
There are a variety of adverse effects and special population warnings that apply across the class of antipsychotic medications. These should be taken into account when selecting an agent, structuring the treatment plan, and conducting follow-up assessments.
In general, the management of behavioral and psychological symptoms of dementia (most prominently psychosis and agitation/aggression) relies on the off-label use of atypical antipsychotics, along with any effective behavioral interventions. Unfortunately, these drugs are often less than clinically effective, with many patients displaying no or only partial response [26]. Furthermore, elderly patients with dementia are much more likely to experience the adverse effects of antipsychotics. The FDA has included a boxed warning on the labels of all atypical antipsychotics regarding the increased mortality risk when these medications are used in elderly patients [26]. The APA recommends that nonemergency antipsychotic medication should "only be used for the treatment of agitation or psychosis in patients with dementia when symptoms are severe, are dangerous, and/or cause significant distress to the patient" [27].
Some antipsychotics are approved or used for the treatment of depressive episodes in bipolar disorder or as adjunctive treatment for unipolar depression. In this capacity, they are considered antidepressants, and antidepressants have been shown to increase the risk of suicidal thinking and behavior in children, adolescents, and young adults (≤24 years of age) in the first months of treatment [28]. As such, a boxed warning has been added to the labeling information for these agents regarding this increased risk. This risk will be discussed in more detail later in this course.
Neuroleptic malignant syndrome is a life-threatening idiosyncratic reaction to antipsychotic medications (including newer atypical agents, although it is more common with typical antipsychotics). It is a rare condition (occurring in approximately 0.01% to 0.02% of patients receiving antipsychotics), but it is unpredictable and potentially fatal, making it a significant concern [29]. Higher doses, higher potency, and long-acting formulations increase the risk of neuroleptic malignant syndrome.
Onset of symptoms is generally apparent within two weeks of treatment initiation. Clinical presentation consists of rigidity/stiffness, high fever, sweating, confusion, unstable blood pressure, and agitation. Signs and symptoms usually progress to a peak after three days. Prompt recognition is necessary to avoid significant morbidity and mortality, and treatment is immediate cessation of the antipsychotic medication and implementation of supportive measures (e.g., hydration, cooling) [30]. In more severe cases, administration of bromocriptine mesylate or dantrolene sodium may be indicated.
An often-overlooked side effect of antipsychotics is elevated prolactin levels, which can present in men as gynecomastia or in women as galactorrhea and absence of menses. Elevated prolactin levels should be monitored and may necessitate changing agents.
As noted, extrapyramidal reactions can occur with antipsychotic therapy and can be a significant barrier to the effective use of these agents. There have also been anecdotal reports of dyskinesia seen with virtually all classes of medications, but the majority of patients with tardive dyskinesia have had exposure to antipsychotics. Studies estimate that up to 30% of patients on long-term antipsychotic therapy will develop various movement disorders [17,31]. The risk is also elevated in elderly patients.
The pathophysiology of tardive dyskinesia is believed to be related to abnormal functioning of the extrapyramidal tracks in the nervous system. Tardive literally means "late occurring," and although there are case reports of patients having symptoms after a single dose of a neuroleptic medication, the primary defining factor of tardive dyskinesia is that it is a later-occurring syndrome, as opposed to dystonia and akathisia, which usually develop in the early phases of treatment.
Tardive dyskinesia is also characterized by involuntary, uncontrollable movements. This differentiates it from dystonia, which is characterized by stiffness and decreased movement and can often be identified during AIMS testing by flexing the patient's wrists and elbows and observing for resistance. Akathisia is the perception of wanting to move and difficulty staying still; it often manifests as a constant need to pace or walk. In AIMS testing, akathisia can be identified in patients who have difficulty sitting without tapping feet, moving their legs, or otherwise having difficulties sitting still. Tardive dystonia typically presents with a fixed posturing of the face and neck, often a sideways tilt of the head, that the patient is unable to voluntarily correct. The most classic presentation of this is torticollis.
Proper diagnosis of movement disorders is essential, as treatment should be tailored to the specific disorder. Work-up of movement disorders should include consultation with appropriate specialists. This is especially important in the treatment of elderly patients for which the onset of parkinsonian movements may in fact be the hallmark of primary Parkinson disease as opposed to secondary parkinsonian side effects. Further assessment should be guided by neurologic consultation and may involve imaging.
After the specific movement disorder has been identified, treatment will focus on the goals of decreasing the movement disorder and the need to avoid destabilizing the patient's psychotic disorders.
Treatment of Tardive Dyskinesias
Treatment of tardive dyskinesia can be difficult and an impediment to the clinical stabilization of the patient. The primary consideration for patients receiving typical antipsychotics should be dose reduction or switching medications. Patients should be warned that tardive dyskinesia symptoms may initially worsen transiently as medication dosages are lowered (i.e., withdrawal-emergent dyskinesias). If a new medication will be used, clozapine should be considered first, as it has the lowest risk of tardive dyskinesia. However, it is vital to ensure that control of psychosis is maintained, particularly in patients at risk for self-harm or violence.
In 2017, the FDA approved the first medications for the treatment of tardive dyskinesia: valbenazine (Ingrezza) and deutetrabenazine (Austedo). The initial dose of valbenazine is 40 mg once daily. This is increased to 80 mg once daily after one week [19]. Continuation of a daily dose of 40 mg or 60 mg may be considered based on response and tolerability. Improvement is seen in 2 to 6 weeks, with the result stabilizing between 16 to 32 weeks. Deutetrabenazine is started at a dosage of 6 mg twice daily. This is increased as needed and tolerated in increments of 6 mg/day up to a maximum of 48 mg/day [19].
Although there is little evidence supporting this use, anticholinergics are often used to prevent extrapyramidal side effects caused by antipsychotics. Some clinicians will co-prescribe these drugs in an effort to prevent the development of extrapyramidal reactions. However, studies have shown that the numerous side effects associated with anticholinergics (e.g., cognitive impairment, urinary disturbances, vision changes, constipation) may preclude their use [31,32]. Elderly patients are particularly vulnerable to these effects, and male patients older than 40 years of age are especially susceptible to acute urinary retention. The Canadian Psychiatric Association recommends that clinicians consider discontinuation of anticholinergic medications for the treatment of tardive dyskinesia, keeping in mind that there is very little evidence to support this course of action and that drug-induced parkinsonism may worsen [33]. If anticholinergics are used, they should be prescribed at the lowest possible dose, patients should receive education on the potential risks and benefits, and informed consent should be obtained.
As discussed, risk mitigation strategies should be initiated when prescribing any antipsychotics. Although only clozapine has a formal REMS program, there are steps that should be taken with any antipsychotic prescription to minimize the potential for adverse effects and manage associated risks (Table 4).
RISK MITIGATION STRATEGIES FOR PATIENTS PRESCRIBED ANTIPSYCHOTICS
| Risk Mitigation Strategy | Frequency of Assessmenta | ||
|---|---|---|---|
| AIMS testing | Every three months and after each dosage change | ||
| Electrocardiogram |
| ||
| Weight | At each visit | ||
| Vital signs | At each visit | ||
| Laboratory studiesb |
| ||
| |||
Patients prescribed antipsychotics should receive education on the effects of the drugs, possible risks and benefits, and any recommended monitoring. However, this can be difficult if the patient has active psychosis or delusional paranoia. In these cases, caregivers and/or surrogate decision makers should be sought. All discussions should be fully documented. Patient education is an ongoing process (not a single event), and efficacy and adverse effects should be discussed regularly with patients as well as steps taken to treat or ameliorate adverse effects.
While Table 4 presents an example of risk mitigation strategies for all antipsychotics, the actual strategies used should be specific to the patient and agent(s); abnormalities should generate a consult to appropriate medical team members. Aside from prevention and early detection of adverse effects, risk mitigation strategies should include a treatment plan for any possible adverse reactions. As discussed, there are several class-wide potential adverse effects, and these known risks should be reviewed and planned for.
Patient A is 34 years of age with a history of schizoaffective bipolar disorder, first diagnosed at 18 years of age during his first year at college. His childhood was notable for several episodes of brief counseling for behavioral issues in school and at home, with a brief trial of methylphenidate (Ritalin) at 11 years of age. Patient A graduated from high school with his peers, but once at college, he developed delirious mania, paranoia, and psychosis. Treatment with olanzapine and valproic acid (Depakote) was initiated, but compliance has been an ongoing issue. During the past 16 years, the patient has had numerous episodes requiring hospitalization, one suicide attempt, and three arrests for breach of peace. He is now on probation and has been unable to maintain employment. He is living with his mother, who has communicated that he will have to leave the house if he does not take his medication. Patient A presents for medication management, stating that he does not like the weight gain he has experienced with many of the medications he has used in the past.
The nurse takes and documents a full history. On mental status exam, Patient A is alert and oriented. He scores 27 on a Mini-Mental State Exam (MMSE) and is mildly grandiose. His speech is slightly pressured but interruptible. At the time of presentation, Patient A is not taking any medications, having disagreed with his last provider's prescription of olanzapine, a drug that had been effective in the past.
Patient A is 6 feet 1 inch tall and weighs 235 pounds. His blood pressure is 124/86 mm Hg. Laboratory studies are requested, and all return normal. An EKG reveals a normal QTc and sinus rhythm.
The nurse asks Patient A what he wants to gain in his medication management, and his stated goals are to continue to live with his mother, to avoid future arrest, and to be able to maintain a job. The nurse discusses available typical and atypical oral and decanoate antipsychotics. After a long discussion, Patient A decides to try long-acting injection risperidone (Risperdal Consta). Patient education includes the risks of extrapyramidal reactions (e.g., movement disorders), weight gain, and prolactin elevation, which can lead to gynecomastia, as well as risk mitigation strategies if any of these develop.
A baseline AIMS test is conducted and is negative for any existing issues. The oral trial of risperidone is initiated, as required before using any long-acting injectable to demonstrate tolerance. The patient is titrated up to an oral dosage of 3 mg twice daily, which appears to resolve the patient's paranoia, pressured speech, and hypomania. Patient A reports that he is functioning better, but he reiterates that he does not think he will be able to comply with daily oral medication. The decision is made to progress to long-acting injected risperidone, which requires a three-week overlap with the oral medication.
While there is no exact dosage equivalency, the manufacturer recommends that a 6-mg total oral daily dose of risperidone should be converted to a bimonthly injectable dose of 37.5–50 mg long-acting risperidone. The patient is started at 37.5 mg IM every two weeks. After three weeks, the patient is stable and able to wean off the oral risperidone, decreasing to 4 mg in two divided doses for three days, then 2 mg in divided doses for three days, before total cessation. During this time, the nurse maintains frequent contact with Patient A to verify stability and compliance.
After six weeks, laboratory studies are ordered, including blood glucose level, lipids, and prolactin level, and a repeat EKG is done. All findings are normal. The nurse continues to administer the AIMS test every three months and to check weight and vital signs at every visit. While Patient A continues with this therapy, laboratory values are rechecked every three to six months.
Patient A responds well to treatment and is able to remain living with his mother. After six months, he has gained employment at a fast-food restaurant and has avoided any contact with law enforcement.
Mood disorders affect approximately 9.7% of the U.S. population each year [34,35,36]. At least one major depressive episode occurs in approximately 21 million American adults, or about 8.3% of the U.S. population 18 years of age and older in a given year. The lifetime incidence of depression in the United States is 20% in women and 13% in men, or about 17% of all Americans.
There are five major classes of antidepressants available in the United States: tricyclic antidepressants (TCAs), MAOIs, SSRIs, serotonin-noradrenaline reuptake inhibitors (SNRIs), and atypical antidepressants; in 2023, the FDA approved gepirone, the first of a new class of antidepressants referred to as serotonin 5-HT1A receptor agonists (Table 5). Modern antidepressants were introduced in the 1950s following serendipitous discovery of antidepressant effects with TCAs and MAOIs. SSRIs were introduced in the late 1980s, followed by atypical antidepressants and SNRIs [37]. The monoamine hypothesis, proposed to explain the unexpected effects of TCAs/MAOIs in the 1950s, posits that depression results from deficient brain serotonin and/or norepinephrine levels. This remained the dominant paradigm of depression and the basis of nearly all FDA-approved antidepressants for the next five decades [38,39]. However, limitations of the monoamine hypothesis and mechanistic homogeneity of standard antidepressants are now understood. Major depressive disorder is more complex and diverse than previously assumed, and novel pathways that underlie its pathophysiology have been identified [40,41].
ANTIDEPRESSANT MEDICATIONSa
| Drug | Dose Range | Typical Starting Dose | Potential Adverse Effects | Comments | ||||
|---|---|---|---|---|---|---|---|---|
| MAOIs | ||||||||
| Selegiline (Emsam, Zelapar) | 6–12 mg transdermal patch every 24 hours | 6 mg transdermal patch every 24 hours |
| Also used in treatment of Parkinson disease | ||||
| Isocarboxazid (Marplan) | 10–40 mg/day | 10 mg BID | May take 3 to 6 weeks to see effects. Dose should be reduced once maximum clinical effect is seen. If no response obtained within 6 weeks, additional titration is unlikely to be beneficial. | |||||
| Phenelzine (Nardil) | 15–30 mg every 8 hours | 15 mg every 8 hours | — | |||||
| Tranylcypromine (Parnate) | 10–60 mg BID | 10–30 mg BID | — | |||||
| Moclobemide | 300–600 mg/day | 300 mg/day in 2 divided doses | — | |||||
| Tricyclic Antidepressants | ||||||||
| Amitriptyline | 50–300 mg/day | 25–50 mg/day as a single dose at bedtime or in divided doses | Xerostomia, sedation | Follow levels and EKG/QTc | ||||
| Clomipramine (Anafranil) | 12.5–250 mg at night | 12.5–50 mg at night | Approved for OCD, off-label for MDD | |||||
| Doxepin (Silenor) | 25–300 mg at night or in divided doses | 25–50 mg at night | Usually reserved for treatment-resistant MDD | |||||
| Imipramine | 25–300 mg at night or in divided doses | 25–50 mg at night or in divided doses | — | |||||
| Trimipramine | 25–300 mg at night or in divided doses | 25–50 mg at night or in divided doses | — | |||||
| Amoxapine | 25–600 mg total (may be BID dosing) | 25–50 mg at bedtime or BID | The maximum dose in outpatients is 400 mg/day; in hospitalized patients, it is 600 mg/day. | |||||
| Desipramine (Norpramin) | 25–300 mg daily or in divided doses | 25–50 mg/day | — | |||||
| Nortriptyline (Pamelor) | 25–150 mg/day | 25 mg at night | — | |||||
| Protriptyline | 10–60 mg daily divided in 3 to 4 doses | 10–20 mg daily divided in 3 to 4 doses | — | |||||
| SSRIs | ||||||||
| Citalopram (Celexa) | 20–40 mg/day | 20 mg/day | GI upset |
| ||||
| Escitalopram (Lexapro) | 10–20 mg/day | 10 mg/day | GI upset | Also approved for generalized anxiety disorder | ||||
| Fluoxetine (Prozac) | 20–80 mg/day | 20 mg/day | GI upset, activation syndrome | Also approved for bulimia, panic disorder, generalized anxiety disorder, OCD, and PMDD | ||||
| Fluvoxamine, immediate-release | 50–300 mg at night | 50 mg at night | Nausea | Approved for OCD, off-label for MDD | ||||
| Fluvoxamine, controlled-release | 100–300 mg at night | 100 mg at night | ||||||
| Paroxetine (Brisdelle, Paxil, Pexeva) | 20–50 mg/day | 20 mg/day | Sedation | Also approved for generalized anxiety disorder, panic disorder, OCD, PTSD, PMDD, social anxiety disorder, and vasomotor symptoms of menopause | ||||
| Paroxetine, controlled-release (Paxil CR) | 25–62.5 mg/day | 25 mg/day | Sedation | |||||
| Sertraline (Zoloft) | 50–200 mg/day | 50 mg/day | GI upset | Also approved for OCD, panic disorder, PMDD, PTSD, and social anxiety disorder | ||||
| SNRIs | ||||||||
| Venlafaxine (Effexor) | 37.5–375 mg BID | 37.5–75 mg BID | Nausea, hypertension, xerostomia, drowsiness | Use with caution in patients with glaucoma | ||||
| Venlafaxine, extended-release (Effexor XR) | 37.5–225 mg/day | 37.5–75 mg/day | Nausea, xerostomia, hypertension |
| ||||
| Levomilnacipran (Fetzima) | 20–120 mg/day | 20 mg/day | Orthostatic hypotension (dose related), nausea | — | ||||
| Desvenlafaxine (Pristiq) | 50–100 mg/day | 25-50 mg/day | Dizziness, insomnia, hyperhidrosis, nausea, xerostomia, anxiety |
| ||||
| Duloxetine (Cymbalta, Drizalma Sprinkle) | 40–120 mg/day | 40–60 mg/day | Activation syndrome, weight loss, GI upset, headache |
| ||||
| Milnacipran (Savella) | 25–100 mg BID | 25–50 mg BID | Nausea, headache, constipation, insomnia | Approved for fibromyalgia, off-label for MDD | ||||
| Atypical Antidepressants | ||||||||
| Bupropion (Aplenzin) | 75–450 mg/day in divided doses | 100 mg BID | Increased seizure threshold, weight loss, GI upset, agitation |
| ||||
| Bupropion, sustained-release (Wellbutrin SR) | 150–200 mg BID | 150 mg/day in the morning | ||||||
| Bupropion, extended-release (Forfivo XL, Wellbutrin XL) | 150–450 mg/day in the morning | 150–175 mg/day in the morning | ||||||
| Mirtazapine (Remeron) | 15–45 mg at bedtime | 15 mg at bedtime | Weight gain, increased appetite, drowsiness | Weight gain may limit satisfaction and compliance | ||||
| Trazodone | 50–600 mg BID | 50 mg BID | Drowsiness, dizziness, xerostomia, GI upset | — | ||||
| Nefazodone | 50–600 mg in divided doses | 50–100 mg BID | Headache, xerostomia, drowsiness | Should not be initiated in individuals with active liver disease or elevated baseline serum transaminases | ||||
| Brexanolone (Zulresso) | — |
| Drowsiness, sedation, xerostomia, dizziness |
| ||||
| Esketamine (Spravato) | 56–84 mg intranasal twice weekly | 56–84 mg intranasal twice weekly | Dissociation, anxiety, nausea, dizziness |
| ||||
| Serotonin 5-HT1A Receptor Agonist | ||||||||
| Gepirone (Exxua) | 18.2–72.6 mg | 18.2 mg/day | Nausea, dizziness | No sexual side effects or weight gain | ||||
| Multimodal Agents | ||||||||
| Vilazodone (Viibryd) | 10–40 mg/day | 10 mg/day | Headache, GI upset | Alternative agent | ||||
| Vortioxetine (Trintellix) | 5–20 mg/day | 5–10 mg/day | Nausea, sexual dysfunction | |||||
| ||||||||
Unlike psychotic disorders, first-line therapy for mild depression should include nonpharmacologic interventions, such as cognitive behavioral therapy, guided self-help, interpersonal psychotherapy, and other lifestyle and psychosocial interventions. Evidence-based guidelines recommend that the process of treatment selection involve shared decision making between provider, patient, and family members, and that the values, priorities, and goals of the patient be included in discussions of risks and benefits of treatment options [42]. Ongoing communication with other providers involved in the care of a patient is essential for coordination and monitoring and can involve different care providers in the same primary care clinic or the primary care provider and therapist or psychiatrist [43].
Combining pharmacotherapy and psychotherapy treatments should be considered for patients with major depressive disorder when practical, feasible, available, and affordable. Both approaches combined show better outcomes than either as monotherapy. When unable to combine therapy because of patient preference or problems with availability or affordability, consider psychotherapy when the presentation is mild-to-moderate, and antidepressants when depression is severe or chronic. Patients with major depressive disorder with psychotic features should receive an antipsychotic and an antidepressant medication or ECT. Lithium can be added in patients unresponsive to antipsychotic/antidepressant therapy [43]. Other specific factors should be considered in treatment planning, including the presence of substance abuse, specific features, and other comorbid disorders. If a patient displays signs of potential suicide, increasing the treatment intensity, including hospitalization if needed, should be considered, and pharmacotherapy and psychotherapy should both be provided [43].
Prior to the prescription of antidepressants, patients should have a full history and physical exam and any relevant medical conditions (e.g., heart failure, renal disease) should be noted. A full medication reconciliation should be conducted, with special attention to potential interactions.
Treatment with antidepressant medications can involve dosage adjustments and/or trials of a different medication at some point to maximize response and minimize side effects [43]. Patient adherence to the medication regimen is essential in achieving the maximum clinical benefit. Providers should closely monitor patients for worsening depressive symptoms and emergent suicidality; appropriate intervention includes stopping or modifying the drug therapy or hospital admission [44]. Providers should instruct patients and caregiver(s) to be alert for emerging agitation/irritability, suicidality, and worsening depression, and to report this immediately to a healthcare provider [44].
The treatment efficacy of antidepressants in major depressive disorder is broadly similar. Other factors to help guide medication selection include previous patient or family member response to antidepressants (if any); impact on psychiatric or medical comorbidities; clinician familiarity; patient preference; safety in overdose; availability and cost; and drug-drug interactions [44]. Most second-generation antidepressant drugs are recommended as first-line treatment due to the quality of published data, side effect tolerability, and safety in overdose relative to TCAs and MAOIs [43,44,45].
The three most distressing side effects for patients treated with antidepressants are sleep disturbance, sexual dysfunction, and weight gain [46]. Choice of medication should be guided by knowledge of comparative side effects and patient priorities; some patients will be more concerned about sexual side effects, while for others, nausea, sleep disturbances, or weight gain may be more distressing [47]. In addition, available evidence regarding the optimal pharmacotherapeutic selection for the treatment of dimensions of depression and DSM-5-TR specifiers should be considered.
SSRIs are thought to act by inhibiting serotonin transporters (SERT) that reuptake serotonin (5-HT) into the presynaptic cell, increasing 5-HT in the synaptic cleft. SSRIs have advantages of low overdose lethality and better tolerability than first-generation antidepressants, which can improve adherence. SSRIs are particularly effective in patients with obsessive-compulsive symptoms, but may initially worsen anxiety or panic symptoms [35,43,44]. This class includes the agents fluoxetine (Prozac), paroxetine (Paxil), sertraline (Zoloft), fluvoxamine (Luvox), citalopram (Celexa), escitalopram (Lexapro), and vortioxetine (Brintellix). Escitalopram may have fewer drug-drug interactions than other SSRIs, and fluoxetine may be a better choice in patients with poorer adherence due to its long half-life [35,43,44].
The most common side effects with SSRIs are gastrointestinal (nausea, vomiting, and diarrhea), activation/insomnia (restlessness, agitation, anxiety, akathisia, and sleep disturbances), sexual, headache, fatigue, and weight gain [35,43,44]. Many of these side effects dissipate over time. Sertraline is particularly associated with diarrhea, and paroxetine with weight gain [35,43].
SNRIs act by inhibiting the reuptake of the neurotransmitters serotonin and norepinephrine. This results in an increase in the extracellular concentrations of serotonin and norepinephrine and therefore an increase in neurotransmission [35,43,44]. Most SNRIs, including venlafaxine (Effexor), desvenlafaxine (Pristiq), levomilnacipran (Fetzima), and duloxetine (Cymbalta), are several-fold more selective for serotonin than norepinephrine.
Safety, tolerability, and side effect profiles of SNRIs resemble SSRIs, with the exception that the SNRIs have been associated (rarely) with sustained elevated blood pressure. SNRIs can be used as first-line agents, particularly in patients with significant fatigue or comorbid chronic pain, and have an important role as second-line agents in patients who have not responded to SSRIs [35,43,44].
Venlafaxine is especially beneficial in treating anxiety and panic attacks in patients with depression, and acts like an SSRI at lower doses (75 mg/day) but more like an SNRI at doses ≥150 mg/day [35,43,44].
TCAs are predominantly serotonin and/or norepinephrine reuptake inhibitors that act by blocking the serotonin transporter and the norepinephrine transporter, respectively, which results in an elevation of the extracellular concentrations of these neurotransmitters, and therefore an enhancement of neurotransmission. TCAs also have varying but typically high affinity for the H1 and H2 histamine receptors and muscarinic acetylcholine receptors. As a result, they also act as potent antihistamines and anticholinergics. These properties are generally undesirable in antidepressants, however, and likely contribute to their large side effect profiles [48].
TCAs are classified by the nature of the final amine group on the side chain, with the tertiary amines amitriptyline (Elavil), clomipramine (Anafranil), doxepin (Sinequan), trimipramine (Surmontil), imipramine (Tofranil), and lofepramine (Lomont); the secondary amines nortriptyline (Pamelor), desipramine (Norpramin), and protriptyline (Vivactil); and the tetracyclic antidepressants amoxapine (Asendin) and maprotiline (Ludiomil).
TCAs are comparable in efficacy to SSRIs/SNRIs, but their side effect profile makes them seldom used as first-line therapy [35,43,44]. TCAs may initially worsen anxiety or panic symptoms. Due to side effect potential of cardiac arrhythmia, TCAs should be used very cautiously, if at all, in patients with heart problems. Secondary amine TCAs cause less orthostatic hypotension and sedation than tertiary amines, which should be avoided in elderly patients due to the risk for orthostatic hypotension, sedation, cognitive problems, and cardiac effects. The secondary amine nortriptyline is especially effective for elderly patients with moderate-to-severe depression. Clomipramine is particularly effective in patients with obsessive-compulsive symptoms [35,43,44].
Anticholinergic and antihistamine activity accounts for many side effects, including dry mouth, blurred vision, reduced gastrointestinal motility or constipation, urinary retention, cognitive and/or memory impairment, and increased body temperature [35,43,44]. Other side effects may include drowsiness, anxiety, emotional blunting (apathy/anhedonia), confusion, restlessness, dizziness, akathisia, hypersensitivity, changes in appetite and weight, sweating, sexual dysfunction, muscle twitches, weakness, nausea and vomiting, hypotension, tachycardia, and arrhythmia. Tolerance to side effects often occurs if treatment is continued. Side effects may also be less troublesome if treatment is initiated with low doses and gradually increased [48,49].
MAOIs inhibit monoamine oxidase (MAO), an enzyme that degrades and inactivates 5-HT, norepinephrine, and dopamine. This increases monoamine levels and activity. Earlier MAOIs were irreversible MAO inhibitors, deactivating the enzyme until slowly replenished over a two-week period [50,51]. MAOIs includes phenelzine (Nardil), tranylcypromine (Parnate), isocarboxazid (Marplan), linezolid (Zyvox, Zyvoxam, Zyvoxid), moclobemide (Aurorix, Manerix), pirlindole (Pirazidol) (approved for use in parts of Europe), and selegiline (Deprenyl, Eldepryl, Emsam).
Most MAOIs appear broadly effective in a range of depressive and anxiety disorders, and may be more effective than other antidepressant classes in major depressive disorder with pronounced anxiety or panic symptoms [35,43,44]. Major depressive disorder with atypical features may preferentially respond to MAOIs over other antidepressant classes. Although effective, MAOIs are rarely the first- or second-line treatment choice due to serious side effect potential from medication interactions and dietary restriction. The selegiline transdermal patch (Emsam) used at the lowest strength (6 mg delivered over 24 hours) may lack the dietary restrictions required of oral MAOIs [52].
With oral ingestion, MAOIs inhibit the catabolism of dietary amines. When foods containing tyramine are consumed, the individual may suffer from hypertensive crisis [50]. If foods containing tryptophan are consumed, hyperserotonemia may result. The amount required to cause a reaction varies greatly from individual to individual and depends on the degree of inhibition, which in turn depends on dosage and selectivity.
MAOIs should not be combined with psychotropic drugs or with any other psychoactive substance except under expert care. This includes a wide range of prescribed, over-the-counter, and illicit drugs and nutritional supplements, such as St. John's wort. Common side effects include orthostatic hypotension, weight gain, sexual dysfunction, sedation, headache, and insomnia [50].
The atypical antidepressants are diverse in monoamine activity and do not fit the profile of other classes. They include bupropion (Wellbutrin), nefazodone (Serzone), mirtazapine (Remeron), and trazodone (Desyrel). Nefazodone and trazodone block postsynaptic serotonin type-2 receptors and inhibit presynaptic serotonin reuptake. Bupropion inhibits activity of norepinephrine and dopamine transporters, and the active metabolite hydroxybupropion contributes to the drug's effects. Mirtazapine is a potent antagonist at 5-HT2, 5-HT3, alpha2, and H1 histamine receptors. As a group, these agents show low toxicity in overdose and may have an advantage over the SSRIs by causing less sexual dysfunction and gastrointestinal distress [35,43,44].
Each agent has apparent benefits and drawbacks, with some better suited for specific patient populations. Bupropion is associated with a risk of seizure at higher doses, especially in patients with a history of seizure or eating disorders, and should be used cautiously in anxious patients [35,43,44]. It may be more effective for atypical major depressive disorder than other antidepressants.
Mirtazapine can be very sedating and promotes appetite and weight increase, which in some patients may be desirable. It has a faster onset of action than fluoxetine, paroxetine, or sertraline, and may be superior to SSRIs in depression associated with severe insomnia and anxiety. Trazodone is also very sedating and is usually used as a sleep aid rather than as an antidepressant [35,43,44].
The side effects of atypical antidepressants vary considerably. With bupropion, the most common side effects are agitation, jitteriness, mild cognitive dysfunction, insomnia, gastrointestinal upset, and possible increased risk for seizures [35]. Patients taking mirtazapine may experience dry mouth, sedation, weight gain, and increased serum cholesterol [43]. Sedation is the most common side effect associated with trazodone, followed by cardiovascular side effects (such as orthostasis) and sexual side effects [43]. Finally, nefazodone is associated with sedation, dry mouth, nausea, constipation, orthostasis, visual alterations, and possible increased risk of hepatotoxicity; this has led to nefazodone being seldom prescribed [35].
Gepirone is the first and only selective 5-HT1A receptor partial agonist approved by the FDA for the treatment of major depressive disorder [19]. Gepirone is in the azapirone group of compounds related to buspirone (which is approved for generalized anxiety disorder). It is noted for its absence of sexual side effects and weight gain and is being studied as a treatment for hypoactive sexual desire disorder in men and women [131,132]. It is hypothesized that the libido improvement seen in studies was independent of its antidepressant or antianxiety effects. The most common side effects are nausea and dizziness, but, according to the manufacturer, these are usually mild, transient, and associated with dosage increases [133].
Vilazodone and vortioxetine are multimodal antidepressants that combine SSRI properties with other pharmacologic actions affecting monoamine and non-monoaminergic targets. Evidence does not suggest greater efficacy than SSRI/SNRIs, but these agents may improve tolerability or efficacy on specific clinical domains [53].
Vilazodone, approved in 2011, primarily acts as a SERT inhibitor and 5-HT1A receptor partial agonist, and modestly inhibits dopamine and norepinephrine transporters. This antidepressant may be most helpful in patients lacking response to initial SSRIs. Vilazodone must be taken with food, which increases its absorption and bioavailability by 72% [54,55].
Vortioxetine, approved in 2013, acts through various serotonin receptors as an antagonist (5-HT3/7/1D), partial agonist (5-HT1B), or agonist (5-HT1A), and inhibits SERT. It also activates the glutamate system in the frontal cortex. Vortioxetine displays a specific clinical efficacy in the treatment of cognitive deficits associated with major depressive disorder. The most common side effects are nausea, vomiting, and constipation [53].
Nearly all commercially available antidepressants are associated with sexual side effects. SSRI/SNRIs show the highest rates of sexual dysfunction, including impaired sexual motivation, desire, arousal, and orgasm affecting men and women. Prescribers greatly underestimate the prevalence and patient burden of sexual side effects from antidepressants and other medications [56]. Among antidepressants, prevalence rates of sexual side effects are highest with venlafaxine and SSRIs; moderate with TCAs and MAOIs; low with bupropion, trazodone, nefazodone, mirtazapine, agomelatine, and vilazodone; and lowest with the reversible MAOI moclobemide [57,58]. As noted, gepirone is not associated with sexual dysfunction. Compared to spontaneous patient reporting, systematic inquiry increases the rate of identifying sexual side effects by ≥60% [58].
Management of sexual side effects in men includes the use of phosphodiesterase-5 inhibitors such as sildenafil, vardenafil, tadalafil, and avanafil as first-line treatment or switching to bupropion [59]. In women, sexual side effect management considers symptoms, age, and potential hormonal contribution when peri- or post-menopausal.
Several papers documenting an increased risk of suicidal thoughts and behavior with antidepressants, primarily SSRIs, have been published over the past decade. A review of the literature found that antidepressant use, including SSRIs, carried a small short-term risk of inducing suicidal thoughts and suicide attempts in persons younger than 25 years of age, with persons 30 to 40 years of age having a lower risk than those younger than 25 years. This risk should be balanced against the well-known beneficial effects of antidepressants that include reduced suicidal ideation and behavior, particularly in the long term. Clinical decision making should weigh the benefits and potential risks and strive to keep the potential risks of antidepressant treatment to a minimum [60,61].
Antidepressant discontinuation (more appropriately termed withdrawal) symptoms are described by the FINISH mnemonic (flu-like symptoms, insomnia, nausea, imbalance, sensory disturbances, hyperarousal), may be experienced by up to 40% of patients when antidepressants are stopped abruptly, and may occur with any antidepressant [62,63,64,65]. SSRI withdrawal symptoms are far more frequent with paroxetine. Common symptoms include dizziness, nausea, headache, confusion, low energy, weakness, sleep disturbance, flu-like symptoms, restlessness, agitation, anxiety, panic, anger, and irritability. Less common and more severe symptoms include electric-shock sensations, vertigo, paresthesia, intensified suicidal ideation, aggression, derealization, depersonalization, and visual/auditory hallucinations. Gradual tapering is a reasonable strategy but does not prevent the onset of SSRI withdrawal [66]. SSRI withdrawal syndrome is least likely with fluoxetine and vortioxetine [62].
Symptoms usually begin within five days of treatment cessation or occasionally during taper or after missed doses [67,68]. Symptoms may be severe enough to interfere with daily functioning, and although a four-week taper is usually suggested, some patients may require longer periods, particularly with paroxetine and venlafaxine [69]. Treatment is pragmatic. If symptoms are mild, reassure the patient that this is a common occurrence and that the symptoms will pass in a few days. If symptoms are severe, reintroduce the original antidepressant or a replacement from the same class with a longer half-life, and taper gradually while monitoring for symptoms. Patients should be emphatically informed that the possible or actual emergence of discontinuation symptoms is not a manifestation of addiction to the antidepressant [70]. SSRI withdrawal can also be approached by switching to a course of fluoxetine, such as 10 mg for several weeks, which is slowly tapered and discontinued [43].
Among the antidepressants, only fluoxetine is approved for use in treating major depressive disorder in pediatric patients. In addition, fluoxetine, sertraline, fluvoxamine, and clomipramine are approved for obsessive-compulsive disorder (OCD) in pediatric patients. Other than these indications, all other use of antidepressants in children is off-label [71]. However, these off-label uses are relatively common, particularly among adolescents and pediatric patients with more severe disease.
Beginning in 2003, data have suggested an increase in attempted and completed suicides in children treated with antidepressants [72]. There is also compelling research indicating that the decrease in prescribing of antidepressant medications to adolescents in the period after 2003, both in the United States and the Netherlands, led to an increase in suicide rate [72]. In the Netherlands, the rate increased by 49% between 2003 and 2005, during which time there was a significant decrease in pediatric SSRI prescriptions.
Data for pediatric efficacy (defined as improvements in depressive symptoms) are strongest for fluoxetine, which is approved for children older than 8 years of age [72]. The best results are achieved with a combination of pharmacotherapy and cognitive-behavioral therapy. Due to the risks and complexities involved in the treatment of children with depression, it is recommended that a multidisciplinary team approach be utilized in the treatment of these patients.
Pediatric patients being treated with antidepressants for any indication should be closely observed for clinical worsening, as well as agitation, irritability, suicidality, and unusual changes in behavior, especially during the initial few months of a course of drug therapy, or at times of dose changes, either increases or decreases. This monitoring should include daily observation by families and caregivers and frequent contact with the physician. It is also recommended that prescriptions for antidepressants be written for the smallest quantity of tablets consistent with good patient management, in order to reduce the risk of overdose [71].
The American Psychiatric Association recommends optimizing the medication dose as a reasonable first step for patients treated with an antidepressant who have not responded fully to treatment if the side effect burden is tolerable and the upper limit of a medication dose has not been reached.
(https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf Last Accessed: September 26, 2024)Strength of Recommendation: II (Recommended with moderate clinical confidence)
Standard antidepressants fail to produce adequate response in 30% to 50% and remission in up to 70% of patients with major depressive disorder [73,74,75]. Partial response, instead of full remission, leaves patients with impairing residual symptoms and high risk of relapse. Each relapse increases symptom severity, decreases treatment response, and heightens risk of treatment-resistant depression [76].
Treatment-resistant depression is a problem increasingly encountered by primary care and mental health providers. Contributors to treatment-resistant depression include illness severity, medical and psychiatric comorbidity, and the limitations of FDA-approved drug options. The definition of treatment resistance lacks consensus, but the most common definition is an inadequate response to two or more antidepressants. This does not consider adjunctive strategies or distinguish patients with partial versus non-response [62,77].
In addition to augmentation strategies, a diverse and growing range of interventions are available as options for treatment-resistant depression [78,79,80]. Most engage novel therapeutic targets.
The limitations of standard antidepressants, frequent treatment resistance, and the paradigm shift in psychiatry away from specific neurotransmitter focus and toward an integrative neural network perspective has prompted the development of novel depression treatment approaches, such as neurostimulation therapy. Repetitive transcranial magnetic stimulation (rTMS) is a nonpharmacologic approach to the treatment of depression approved in 2008, and multiple randomized controlled studies have shown its use to be safe and effective [81]. Patient selection for rTMS is complex and should be done in conjunction with the specialist administering the therapy as well as the patient. Most clinicians refer patients after they have failed to improve on at least one antidepressant.
The use of ECT in the treatment of treatment-resistant depression is long and varied. While there was historical misuse and abuse of this modality, particularly in the unethical care of patients in institutions, it has a proven record of efficacy for patients with severe or refractory depression.
ECT is effective as acute treatment, but multiple treatments are required and many who respond experience symptoms again within six months [82]. ECT generates electrical stimuli for seizure induction through electrodes applied to the scalp, with the patient under general anesthesia and pre-medicated with a muscle relaxant. Clinical outcomes are highly influenced by electrode placements, electrical intensity, and pulse width [83].
As first-line treatment, ECT is used for severe melancholic, catatonic, psychotic, or refractory depression and for patients who refuse to eat or drink, have very high suicide risk or severe distress, pregnant women with severe depression, or who have a previous positive ECT response [47,82,84].
Full ECT response requires at least four to six sessions delivered two to three times per week. Twice weekly ECT requires longer treatment duration, but more than three treatments per week is not recommended due to the greater cognitive side effect risk [83].
Headaches (45%), muscle soreness (20%), and nausea (1% to 25%) during ECT are transient and treated symptomatically; 7% of patients with major depression switch into a manic or mixed state [83]. Most distressing to some patients is loss of autobiographic memory recall, infrequently reported to persist beyond six months [84]. ECT lacks absolute contraindication, but increased safety risk is associated with space-occupying cerebral lesion, increased intracranial pressure, recent cerebral hemorrhage, or aneurysm [83,85].
Ketamine is an N-methyl-D-aspartate receptor (NMDA-R) antagonist that was approved for use as an anesthetic in 1970. Demonstration that a single IV dose in patients with treatment-resistant depression reliably produced rapid, robust antidepressant effects for one week was a breakthrough discovery for research and a turning point for patients for whom all other treatment approaches had failed [86]. The short-term efficacy of ketamine treatment of refractory major depressive disorder and bipolar depression is now established; more than one dozen placebo-controlled trials have shown that patients with refractory unipolar or bipolar depression have significantly greater response, remission, and depressive symptom reduction to single-dose IV ketamine than placebo from 40 minutes through days 10 to 12 post-treatment [87,88]. The approach has become standardized, using a sub-anesthetic dose: 0.5 mg/kg IV over a 40-minute infusion. In a 2015 analysis, ketamine was designated as one of two psychiatric treatments that had the highest potential impact on patient outcomes. This designation was based on the serious unmet need for fast-acting, well-tolerated antidepressants with efficacy in refractory major depression and bipolar depression [40].
In 2019, a ketamine derivative—esketamine—was approved by the FDA as an adjunctive agent for treatment-resistant depression. In 2020, the additional indication for short-term treatment of suicidal thoughts was added. This agent is administered by nasal spray and must be given under the direct supervision of a healthcare provider. Patients should be monitored for adverse effects for at least two hours following administration [19,89]. Due to an increased potential for abuse and misuse, it has an associated REMS program.
Patient B, 46 years of age, presents with a history of mild-to-moderate depression for the past six months. He has been undergoing cognitive-behavioral therapy, and while he has made some gains, he continues to experience depressive symptoms, as documented by a mood map and reports from his therapist. In addition, he is overweight and complains of insomnia.
The nurse does a complete psychiatric history and orders baseline laboratory studies and EKG. The patient reports no history of suicidal ideation and has never been hospitalized. He states that he is functioning well in his job but has been unable to maintain a steady relationship, despite a desire to have a long-term partner.
Patient B's main complaints are depressed mood and insomnia. He does not endorse anxiety or panic attacks and denies any drug or tobacco use. The patient is relatively healthy, has received regular medical care, and does not take any other medications. He has no history of seizure disorder. After a long discussion with the patient, the nurse decides to start a trial of sertraline 50 mg/day and trazodone 25 mg at night.
Patient B returns in two weeks and reports that his sleep has improved and the medications are well tolerated. He agrees to return in four more weeks to determine if the medications are helping his mood, which currently has not improved. During that period, he will continue working with his therapist weekly. The nurse and therapist are in good communication, with an understanding that any changes in symptoms or tolerability should be shared immediately.
After four weeks, Patient B comes back to the office. His mood is still described as depressed by both him and his therapist, so the sertraline dosage is increased to 100 mg/day, with a plan for the patient to return in one month.
When Patient B returns in four weeks, he reports that he is feeling better and has been going out and socializing. However, he is experiencing sexual side effects (erectile dysfunction). Several options are discussed, including transitioning from sertraline to a trial of bupropion or adding a trial of sildenafil. The patient decides that he would like to switch to bupropion, because he does not like the idea of having to take another medication.
After decreasing the dose of sertraline to 50 mg for two weeks, the patient is stable and plans are made to discontinue the sertraline and start bupropion (Wellbutrin XR) 150 mg every morning. When Patient B presents in two weeks, he is happy with the new medication, reporting full resolution of the sexual side effects, but reports some residual depressed mood. The dose of bupropion is increased to 300 mg every morning.
In another four weeks, Patient B's depression is in remission and his sleep quality is significantly improved. The medications remain well-tolerated. The patient also notes that he has lost about 5 pounds, which he attributes to decreased appetite. The patient continues to work with his therapist and agrees to return monthly for at least the next three months for continued monitoring.
When using any psychotropic medication for bipolar disorder, the National Collaborating Centre for Mental Health recommends that clinicians ensure that the person is given information that is suitable for their developmental level about the purpose and likely side effects of treatment including any monitoring that is required, and give them an opportunity to ask questions. The choice of medication is made in collaboration with the person with bipolar disorder, taking into account the carer's views if the person agrees. The overall medication regimen is regularly reviewed so that medications that are not needed after the acute episode are stopped.
(https://www.nice.org.uk/guidance/cg185 Last Accessed: September 27, 2024)Level of Evidence: Expert Opinion/Consensus Statement
Perhaps no diagnosis in psychiatry has undergone such a rapid change in diagnostic standards and pharmacologic management as bipolar disorder in the last 30 years. Intravenous chlorpromazine was first noticed to decrease mania in surgical patients with bipolar disorder. Shortly thereafter, chlorpromazine and other antipsychotics became mainstays in the treatment of primary psychotic disorders. At the same time, the treatment of bipolar disorder was revolutionized by the discovery of lithium.
Lithium is perhaps the oldest drug still in clinical use. While its utilization was formally standardized in 1954, its use in psychiatry dates to the mid-19th century [6]. One of the earliest recorded medical uses of lithium, for the treatment of gout, was in 1847 in London. Lithium continued to be prescribed for the treatment of gout and renal calculi into the 1930s. Psychiatric interest in lithium can be traced to the late 1800s. William Hammond is believed to be the first physician to prescribe lithium for mania (in 1871) [6]. As noted, the United States was the 50th country to approve lithium for clinical use in 1970 [6]. Lithium was originally studied in the prevention of depression and continues to be used off-label as an antidepressant [6,90].
Today, management of bipolar disorder may consist of lithium, anticonvulsants, and/or antipsychotic medications (Table 6). Antianxiety medications may be prescribed short term to improve sleep and manage agitation/anxiety. Lithium is one of the few drugs used in the treatment of bipolar disorder with proven efficacy in the prevention of suicide [79].
MEDICATIONS USED IN THE TREATMENT OF BIPOLAR DISORDER
| Drug | Dose Range | Typical Starting Dose | Potential Adverse Effects | Comments |
|---|---|---|---|---|
| Lithium (Lithobid) | 600–1,800 mg/day in divided doses | 300 mg BID | Xerostomia, tremor, thyroid-stimulating hormone elevation, leukocytosis, nausea | Lithium toxicity is closely related to serum lithium levels and can occur at doses close to therapeutic levels. Monitoring should be available before initiating therapy. |
| Anticonvulsants | ||||
| Valproic acid (Divalproex) | 250–1,500 mg BID based on levels | 250–500 mg PO BID | Weight gain, hair loss, GI upset | Drug levels, complete blood count, and hepatic function should be followed. |
| Carbamazepine (Carbatrol, Epitol, Equetro, Tegretol) | 100–800 mg BID | 100–200 mg BID | Weight gain | Hepatic function, drug level, and CBC should be followed. |
| Lamotrigine (Lamictal, Subvenite) | Titrate to maximum 200 mg/day | 25 mg/day | Nausea, tremor, rash, Stevens Johnson syndrome | — |
| Atypical Antipsychotics | ||||
| Aripiprazole (Abilify) | 2.5–30 mg/day | 2.5–5 mg/day | Weight gain (least likely in this class), extrapyramidal symptoms, constipation, sedation | Available in long-acting IM formulation |
| Asenapine (Saphris, Secuado) | 2.5–10 mg BID | 2.5–5 mg PO BID | Weight gain, extrapyramidal symptoms, constipation, sedation | — |
| Cariprazine (Vraylar) | 1.5–6 mg/day | 1.5 mg/day | Weight gain, extrapyramidal symptoms, constipation, sedation | — |
| Lurasidone (Latuda) | 40–160 mg/day | 40 mg/day | Weight gain, extrapyramidal symptoms, constipation, sedation | — |
| Olanzapine (Zyprexa) | 2.5–30 mg either BID or at night | 2.5 mg/day | Weight gain (very likely), extrapyramidal symptoms, constipation, sedation | — |
| Olanzapine/fluoxetine (Symbyax) | Up to 18 mg/day olanzapine and 75 mg/day fluoxetine | 6 mg/day olanzapine and 25 mg/day fluoxetine | Weight gain, extrapyramidal symptoms, constipation, sedation | Usually given at night |
| Quetiapine (Seroquel) | 50–800 mg/day | 50–100 mg daily or BID | Weight gain, extrapyramidal symptoms, constipation, sedation | Available in extended-release formulation |
| Risperidone (Perseris, Risperdal) | 0.5–6 mg/day (may be given BID) | 0.5–1 mg daily or BID | Weight gain, extrapyramidal symptoms, constipation, sedation, increased prolactin, male gynecomastia | Available in long-acting IM formulation |
| Ziprasidone (Geodon) | 40–80 mg BID | 40 mg BID | Weight gain, extrapyramidal symptoms, constipation, sedation, increased prolactin | Taken with food to improve absorption |
| BID = twice per day, CBC = complete blood count, GI = gastrointestinal, IM = intramuscular, PO = oral. | ||||
Patient C, 23 years of age, presents with a history of increasing mood instability, with periods of depression lasting three to four months followed by periods of increasing disorganization, lack of need for sleep, and risky behaviors (e.g., gambling, shoplifting). After her third shoplifting arrest, Patient C agreed to a plea deal that requires her to seek and comply with psychiatric care.
The clinician conducts a full psychiatric history that is significant for school and employment problems. She attempted outpatient treatment at 16 and 21 years of age, both of which were ended by the patient due to her dislike for medications and their side effects and a perceived "loss of energy" when treatment was initiated with olanzapine during a manic episode at 19 years of age.
The workup reveals a healthy woman, normal laboratory studies, normal EKG, and no medical problems. She is gravida 0 para 0 and has a progestin intrauterine device for birth control. She describes her current mood as depressed, rated 3 on a Likert scale. She describes hypersomnia, sleeping about 10 hours per day. She is currently unemployed and living with her parents. She denies any psychotic symptoms and has never attempted suicide. The patient feels her parents are supportive but firm in stating that she must engage in treatment to continue living with them. She is a nonsmoker and has no known drug use, other than occasional cannabis (less than once per week), which she does not view as problematic. She has genetic loading, with an uncle and a sister who have been diagnosed with bipolar disorder.
Patient C is currently not seeing a therapist or receiving psychotherapy. In discussing goals of care, her main goal is compliance with her probation stipulations. She also indicates that she would like to be employed and to eventually move out of her parents' house. Weight gain is identified as an intolerable side effect of pharmacotherapy; the patient states she would like to lose weight. After much discussion, Patient C agrees to see a social worker for cognitive support and to assist with employment goals.
Given the patient's goals and concerns, antipsychotics are determined to be undesirable. The clinician and patient discuss the risks and benefits of valproic acid and lithium. Patient C states that her uncle did well on lithium and that she would like to try it. In preparation for lithium prescription, the patient's thyroid hormone and calcium levels are reviewed along with kidney function; all are normal. Patient C expresses a strong desire to take pills only once per day and does not think she can remember to take a morning dose. The clinician reviews literature, which indicates that once daily dosing of lithium results in similar efficacy, with lower total doses, decreased renal toxicity, and decreased urinary frequency [91]. The patient is started on controlled-release lithium 450 mg taken at night. Eventually, she is titrated to 900 mg daily, with an average serum lithium level of 0.7 mEq/L.
As part of the patient education process, the clinician emphasizes the need for reliable birth control, as lithium crosses the placenta and is associated with serious fetal malformations following exposure in the first trimester [92]. This conversation is documented fully, including a recommendation that the patient use dual birth control (e.g., add condoms). The need to use a different medication during any planned pregnancy is reviewed.
After 12 months of therapy, Patient C has not experienced any episodes of mania. However, she describes her mood as depressed and complains of amotivation, sadness, hypersomnia, and a 10-pound weight gain. Her lithium level is stable and within the therapeutic range, and the clinician is concerned that raising the lithium dose could increase the risk of toxicity.
To address these issues, bupropion (Wellbutrin XL) 150 mg/day is added to the patient's regimen. Augmentation with low-dose bupropion appears to have the lowest risk of inducing mania among available antidepressants [93]. In six weeks, Patient C reports a gradual improvement in mood, a slight weight loss (2 to 3 pounds), and no signs or symptoms of mania. She continues to meet with her social worker and has attained employment as a front desk clerk at a hotel. She is pursuing her goal of living independently and has had no further contact with law enforcement.
Anxiety disorders are characterized by states of chronic, excessive dread or fear of everyday situations. The fear and avoidance can be life-impairing and disabling. Anxiety disorders result from the interaction of biopsychosocial factors, whereby genetic vulnerability interacts with situations, stress, or trauma to produce clinically significant syndromes. Under the umbrella of anxiety disorders are specific phobia, social anxiety disorder, panic disorder, agoraphobia, generalized anxiety disorder, and separation anxiety disorder.
Each year in the United States, anxiety disorders impact approximately 42 million adults, or 19% of the population [94,95]. The pattern of sex distribution is consistent among anxiety disorders, and the overall female-to-male ratio is approximately 2:1 across all age ranges [96]. Guidelines for the treatment of anxiety disorders typically support combined psychotherapy (e.g., mindfulness, exposure therapy, cognitive therapy) and pharmacotherapy [97].
The first report of antidepressant use in anxiety treatment was published in 1962. In this account, patients with agoraphobia who were given the TCA imipramine showed reductions in panic attacks and improved exposure to feared situations [98]. The benzodiazepine chlordiazepoxide (Librium) was introduced to the U.S. market in 1960. This was followed by diazepam (Valium) in 1963, which became the most prescribed drug in the United States from 1969 to 1982; in 1978, more than 2.3 billion diazepam doses were sold in the United States [99]. Panic disorder was first formalized as a psychiatric disorder in the 1980 DSM-III, and alprazolam (Xanax) became the first FDA-approved drug for panic disorder treatment in 1981, remaining the most-prescribed benzodiazepine to date [100].
In the past two decades, antidepressant drugs have displaced benzodiazepines as the most widely prescribed and recommended anxiety disorder pharmacotherapy (Table 7). Antidepressants are generally recommended as first-line therapy for panic disorder because, unlike benzodiazepines, antidepressants treat comorbid depression and lack abuse risk and potential side effects of excessive sedation, cognitive impairment, and ataxia. All major antidepressant classes are comparably effective, but SSRIs and, increasingly, SNRIs are recommended over TCAs and MAOIs due to better safety and tolerability [101].
MEDICATIONS USED IN THE TREATMENT OF ANXIETY DISORDERS
| Drug | Dose Range | Typical Starting Dose | Potential Adverse Effects | Indication(s) |
|---|---|---|---|---|
| Antidepressants | ||||
| Escitalopram (Lexapro) | 10–20 mg/day | 10 mg/day | Few | GAD |
| Fluoxetine (Prozac) | 10–80 mg/day | 10–20 mg/day | Few | OCD, PD |
| Fluvoxamine | 100–300 mg/day | 100 mg/day | Few | OCD, SP |
| Paroxetine (Brisdelle, Paxil, Pexeva) | 10–50 mg/day | 10–20 mg/day | Few | GAD, OCD, PD, SP |
| Sertraline (Zoloft) | 25–200 mg/day | 25–50 mg/day | Few | OCD, PD, SP |
| Duloxetine (Cymbalta, Drizalma Sprinkle) | 30–120 mg/day | 30–60 mg/day | Hypertension, headache | GAD |
| Venlafaxine (Effexor) | 37.5–225 mg/day | 37.5–75 mg/day | Hypertension, headache | GAD, PD, SP |
| Clomipramine (Anafranil) | 25–250 mg/day | 25 mg/day | QTc prolongation | OCD, PD |
| Doxepin (Silenor) | 25–300 mg/day | 25–50 mg/day | QTc prolongation | NSA |
| Imipramine | 50–200 mg/day | 50–100 mg | QTc prolongation | PD |
| Phenelzine (Nardil) | 45–90 mg/day | 45 mg/day | Drug- and food-drug interactions | PD |
| Benzodiazepines | ||||
| Alprazolam (Xanax) | 0.25–4 mg 2 to 3 times per day | 0.25–1 mg/day | Dependence, rebound anxiety, increased dementia risk, increased risk of opioid overdose with concomitant use | NSA, PD |
| Chlordiazepoxide (Librium) | 5–25 mg 3 to 4 times per day | 5–10 mg/day | NSA | |
| Clonazepam (Klonopin) | 0.25–2 mg 2 times per day | 0.25–1 mg/day | PD | |
| Diazepam (Diastat, Valium, Valtoco) | 2.5–10 mg 2 to 4 times per day | 2.5–5 mg/day | NSA | |
| Lorazepam (Ativan) | 0.5–2 mg given 2 to 3 times per day | 0.5–1 mg/day | NSA | |
| Miscellaneous Agents | ||||
| Hydroxyzine (Vistaril) | 25.5 mg 2 to 4 times per day | 25–50 mg/day | Xerostomia, drowsiness | NSA |
| Buspirone | 10–20 mg 2 to 3 times per day | 10 mg/day | Dizziness, headache | GAD |
| GAD = generalized anxiety disorder, NSA = nonspecific anxiety, OCD = obsessive-compulsive disorder, PD = panic disorder, SP = specific phobia. | ||||
The National Collaborating Centre for Mental Health asserts that all patients prescribed antidepressants for the management of anxiety should be informed that, although the drugs are not associated with tolerance and craving, discontinuation/withdrawal symptoms may occur on stopping or missing doses or, occasionally, on reducing the dose of the drug.
(https://www.nice.org.uk/guidance/cg113 Last Accessed: September 27, 2024)Level of Evidence: Expert Opinion/Consensus Statement
SSRIs are considered first-line therapy for generalized anxiety disorder and panic disorder [101,103]. TCAs have comparable efficacy to SSRIs in panic disorder and generalized anxiety disorder [104,105]. TCAs are lethal in overdose and, compared with SSRIs, have a markedly broader, more problematic, and less tolerable side effect profile [101]. Nonetheless, TCAs may work when first-line agents do not [106]. Also, some patients with panic disorder are sensitive to both beneficial and adverse effects of TCAs, so cannot tolerate imipramine doses >10 mg/day but still experience panic blockade [101].
MAOIs are effective for panic disorder and social anxiety disorder and are thought by some to be superior options for severe, treatment-resistant anxiety disorders. As noted, MAOIs have a substantial side effect profile and impose the greatest safety burden of all antidepressants. Therefore, they are usually reserved as the last treatment option after other drug therapies have failed to achieve remission [107]. Clinicians do not routinely prescribe MAOIs for anxiety disorders, although they are probably not considered often enough in treatment-resistant patients [106].
Since their introduction in the early 1960s, benzodiazepines have been the most prescribed drugs for anxiety over the majority of the past half-century. Although SSRI/SNRI agents have replaced benzodiazepines as the top-prescribed anxiolytics, benzodiazepine prescribing remains common. In 2020, alprazolam, clonazepam, lorazepam, and diazepam were among the top 25 most frequently prescribed psychotropic medications in the United States; alprazolam ranked 9th, clonazepam ranked 12th, lorazepam ranked 16th, and diazepam ranked 23rd [108,109].
Numerous benzodiazepines are available and have similar pharmacodynamic properties and clinical actions; they mainly differ in pharmacokinetic properties (absorption, distribution, metabolism, elimination). Benzodiazepines bind to a specific receptor site in the gamma-aminobutyric acid (GABA) receptor complex. GABA is the primary inhibitory neurotransmitter in the central nervous system, and benzodiazepines cause non-selective GABA-A inhibitory effects throughout the brain that include drowsiness, cognitive impairment, dampening of fear and anxiety, memory impairment, anticonvulsant actions, and impairment of balance, motor control, muscle tone, and coordination. Adverse reactions to alprazolam also include amnesia, aggression, mood changes, and hostility. The newer Z drugs (e.g., zolpidem, zopiclone) have similar actions to benzodiazepines but are marketed for insomnia due to their pharmacokinetic profile, with high doses required for anxiolytic effects. There is evidence the Z drugs share similar risks to benzodiazepines [110,111].
Meta-analyses suggest alprazolam, lorazepam, and diazepam are effective but comparable in generalized anxiety disorder efficacy, while clonazepam shows much greater efficacy in the treatment of panic disorder than alprazolam, lorazepam, and diazepam, which all have modest efficacy [112].
Benzodiazepine treatment of anxiety disorders is controversial. While effective in rapid anxiety reduction, the potential drawbacks with long-term use are substantial. These agents are indicated when potent, short-term anxiolytic effects are necessary to permit infrequent exposure to feared stimuli and potentially severe anxiety, such as airplane travel [103,106,113]. Clonazepam, lorazepam, and alprazolam are effective for short-term use in panic disorder, generalized anxiety disorder, and SAD, but ineffective for, and potentially worsening, comorbid depression [114]. The rapid anxiolytic effects make benzodiazepines highly appealing to patients with anxiety, but aside from this specific context, benzodiazepine prescribing for as-needed use is discouraged [106,115,116]. Benzodiazepines can reinforce pill taking, serve as a safety signal that undermines self-efficacy, and become incorporated into conditioned fear responses; these concerns are heightened with as-needed use. On-demand dosing links pill taking to rapid anxiety reduction, powerfully reinforcing avoidance in anxiety-provoking situations and encouraging longer-term reliance on the drug. This iatrogenic effect also contributes to poor response to cognitive-behavioral therapy.
The current recommended prescribing is for time-dependent use, instead of panic response-dependent use, to minimize the risks [103]. This would also seem to maximize risk of withdrawal syndrome from uninterrupted versus intermittent drug exposure.
Benzodiazepines are also useful in the initial weeks of SSRI/SNRI initiation to rapidly reduce anxiety and possible early anxiogenic medication side effects before the onset of SSRI/SNRI anxiolytic effects [103,106,113]. However, patients may discontinue the antidepressant when co-prescribed a rapidly effective benzodiazepine, believing the benzodiazepine's symptom relief makes the SSRI/SNRI unneeded. Supportive therapy with regular visits or phone contacts may also help patients remain adherent until the delayed onset of antidepressant benefits appears or early antidepressant side effects lessen [117].
Another indication for benzodiazepine use is for the short-term relief (two to four weeks only) of anxiety that is severe, disabling, or subjecting the individual to unacceptable distress. Perhaps the greatest prescribing challenge with benzodiazepines is preventing short-term use from insidiously developing into long-term use. Patients with the most severe anxiety may obtain the greatest relief and become most hesitant to discontinue use [118]. In many cases, clinicians ignore the recommended two- to four-week prescribing limit, mainly because alternative options with superior anxiolytic effects are not available [119]. Clinicians intending to prescribe alprazolam should carefully consider the likelihood that its use will remain restricted to the very short term—a few days to a couple weeks—to see the patient through a crisis [118].
Benzodiazepines may be prescribed to augment SSRI/SNRI therapy for improved response in select patients with significant residual anxiety or non-response. In one study, patients with SAD and sertraline nonresponse after 10 weeks were given sertraline plus clonazepam (≤3 mg/day), venlafaxine (≤225 mg/day), or sertraline plus placebo for 12 weeks. Those with sertraline augmented by clonazepam showed greatest reduction in SAD symptoms and a better overall response rate than comparator groups, although remission rates did not differ significantly [120]. These agents are third- or fourth-line treatment in patients unresponsive or intolerant to other anxiolytic drugs who remain highly symptomatic [103,106,113]. Generally, patients with a history of substance abuse, personality disorder, or chronic pain should not be treated with benzodiazepines because of the high risk for overuse of these medications [113]. While benzodiazepines should usually be reserved for patients lacking response to at least two treatments (i.e., non-response to an SSRI/SNRI and a psychologic treatment), concerns about potential problems in long-term use should not prevent their use in patients with persistent, severe, distressing, and impairing anxiety symptoms [121].
It seems the most appropriate guidance for benzodiazepine prescribing involves occasional, context-specific use or cautious use during SSRI/SNRI initiation [121,122]. Otherwise, benzodiazepines should be reserved for patients lacking response to three or more treatments, such as an SSRI, an SNRI, and a psychologic intervention, who remain highly symptomatic.
While alprazolam remains the most-prescribed benzodiazepine for anxiety disorders, evidence suggests that relative to other benzodiazepines, alprazolam is no more effective and may have specific drawbacks [100]. Alprazolam may have greater potential for dependence than other benzodiazepines due to its rapid onset of anxiolysis and short half-life. With the short half-life, persons prescribed fixed-interval alprazolam (e.g., every six to eight hours) can experience morning withdrawal symptoms following the last nighttime dose. This is frequently mistaken as relapse in anxiety for which the drug was originally prescribed, confirming the continuing need for the drug [118]. The alprazolam product monograph states that such emergence of interdose symptoms reflect insufficient plasma levels, best managed by adding the same dose for four times daily administration (but breakthrough anxiety and alprazolam withdrawal are not differentiated). The document also states that alprazolam treatment of panic disorder differs from sub-syndromal anxiety, in that recommended dosing is as close to around-the-clock as possible, or three or four times per day [123].
Long-term benzodiazepine use can result in added symptoms during stable-dose maintenance, including increasing anxiety and withdrawal-associated symptoms such as perceptual disturbances and paresthesia. This emerging withdrawal syndrome despite ongoing benzodiazepine use is much more likely with highly potent and rapidly eliminated alprazolam or lorazepam and is temporarily alleviated by dose escalation. As craving, dysphoria, and other withdrawal symptoms develop over time between doses, the motivation to continue benzodiazepine use for anxiolysis gradually merges with the need to avoid withdrawal symptoms [124].
Benzodiazepine prescriptions are associated with nonmedical use and the development of benzodiazepine use disorder unrelated to co-occurring drug use or anxiety disorder diagnosis/severity [109]. Acute cognitive-impairing side effects are drowsiness, increased reaction time, ataxia, motor incoordination, and anterograde amnesia. In one study, long-term use of an average 17 mg/day diazepam equivalent led to substantial cognitive decline that did not resolve three months after cessation [125]. Motor vehicle accident risks during benzodiazepine therapy are comparable to driving with a blood alcohol concentration of 0.050% to 0.079% [126]. Hip fracture risk is increased by ≥50% in older persons who take benzodiazepines; with zolpidem, the risk is increased 200% in persons older than 65 years of age [127]. The risk of overdose is particularly great when benzodiazepines are combined with sedative drugs such as opioids or alcohol.
Personality traits associated with long-term use, emotional dependence, and more severe/protracted benzodiazepine withdrawal have been described. Long-term benzodiazepine users often have poor stress coping abilities. Benzodiazepines compensate for these deficits, but their use interferes with learning stress coping strategies, including behavioral therapy for agoraphobia. Passive-dependent personality traits and lack of internal and external stress coping resources increases vulnerability to withdrawal symptoms and motivation for continued use. In these patients, benzodiazepine deprivation renders them unprotected from stress and re-exposes their coping deficits. Chronically anxious people have been found innately hypersensitive to punishing stimuli and punishment; benzodiazepines can be described as "depunishing" drugs [124].
Withdrawal symptoms following benzodiazepine cessation are appropriately concerning and a liability of this drug class that all prescribers should understand. In patients with panic disorder discontinuing alprazolam following 1.5 to 22 months of treatment, 33% to 100% were unable to completely taper [43]. These data did not include the 50% of long-term benzodiazepine users who do not consent to withdrawal studies or who later quit the study. The experience of benzodiazepine withdrawal is known to deter patients from future attempts [128]. An estimated 25% to 76% of patients prescribed benzodiazepines are long-term users. Defining high-dose benzodiazepine varies, but users of high-dose benzodiazepines commonly have comorbid disorders and are unlikely to benefit from current discontinuation and withdrawal strategies that expose them to greater risk of impairment and injury [128].
Despite comparable dosing, patients with panic disorder often show greater difficulty tapering than patients with generalized anxiety disorder. Problems during alprazolam tapering are most severe during the last half of the taper. Patients with panic disorder receiving diazepam or alprazolam had fewer problems during taper of the top 50% of daily dose. However, with abrupt discontinuation of the remaining dose, alprazolam caused significantly more anxiety, relapse, and rebound. This may reflect greater problems withdrawing from short half-life, high-potency benzodiazepines like alprazolam [43].
Patient D, 33 years of age, presents with a chief complaint of anxiety. He describes increasing anxiety symptoms for the last eight months, including persistent worrying that he finds challenging to control, overthinking plans, inability to let go of a worry, difficulty concentrating, fatigue, difficulty falling asleep, irritability, and nervousness. He is employed as a corrections officer with two young children. A medical workup is normal. He has engaged with a therapist for the past three months, and while he has made some gains with cognitive-behavioral therapy, his therapist has suggested a medication consultation to help with his ongoing symptoms.
Upon review of symptoms and consultation with the patient's therapist, a diagnosis of generalized anxiety disorder is confirmed. The clinician discusses goals of care with the patient; he states that he wants to avoid the medication side effects of daytime sedation or impaired concentration due to his job. Available medications are reviewed, and the decision is made to start escitalopram to address the continued symptoms of generalized anxiety. Treatment is initiated at a daily dose of 10 mg. Patient D asks about alprazolam (Xanax), as a friend told him that this medication worked for him. The clinician spends time on patient education explaining that alprazolam has a known risk of addiction and a potential for sedation. The patient agrees to the trial of escitalopram.
When Patient D returns for follow-up in four weeks, he reports improvement in his anxiety symptoms and is continuing his cognitive-behavioral therapy. After three months, the patient indicates that his symptoms are manageable and he has not experienced any adverse effects of the pharmacotherapy.
The American Society of Addiction Medicine recommends that all FDA-approved medications for the treatment of opioid use disorder should be available to all patients. Clinicians should consider the patient's preferences, past treatment history, current state of illness, and treatment setting when deciding between the use of methadone, buprenorphine, and naltrexone.
(https://www.asam.org/Quality-Science/quality/2020-national-practice-guideline Last Accessed: September 27, 2024)Level of Evidence: Expert Opinion/Consensus Statement
Although perhaps the most concerning drug of abuse in the United States today is opioids, substance use disorders (as defined by the DSM-5-TR) are not limited to one specific drug. Even commonly used substances, such as alcohol and nicotine, may initiate use disorders. Substance use disorders are diagnosed based on the presence of at least two of the following criteria in the previous year [129]:
Using the substance in larger amounts
Wanting to cut down use but unable to
Spending large amounts of time involved in procuring and using as well as recovering from use of the substance
Cravings for the substance
Not functioning normally at work, home, or school
Continued use despite harm to relationships
Decreasing other activities, including work or socialization
Continued use despite harm or risk of harm
Continued use despite medical or psychological problems made worse by continued use
Increased tolerance leading to escalating dosages
Withdrawal symptoms
The main stages of substance use disorder treatment are crisis intervention, harm reduction, detoxification/withdrawal, active treatment, and relapse prevention. To this end, a variety of medications have been approved to assist in cessation of the use of opioids, alcohol, and nicotine (Table 8). Some are used for detoxification, and others are used to prevent relapse. Research has shown that medications are most effective when used in conjunction with other therapies.
MEDICATIONS USED IN THE TREATMENT OF SUBSTANCE USE DISORDERS
| Drug | Dose Range | Typical Starting Dose | Potential Adverse Effects | Route(s) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Opioid Use Disorder | ||||||||||||||
| Buprenorphine/naloxone (Bunavail, Suboxone, Zubsolv) |
| 4/1 mg/day | Pain, headache, nausea, diaphoresis | Buccal film, sublingual film, sublingual tablet | ||||||||||
| Methadone (Dolophine, Methadose, DISKETS) | 20–120 mg/day | 20–30 mg/day | Pruritus, constipation, cardiac abnormalities | PO, IV | ||||||||||
| Naltrexone (Vivitrol) |
|
| Injection site reactions, anxiety, syncope | PO, IM | ||||||||||
| Buprenorphine (Belbuca, Buprenex, Butrans, Probuphine, Sublocade) |
|
| Few | Sublingual tablet, subdermal implant, SQ injection | ||||||||||
| Alcohol Use Disorder | ||||||||||||||
| Acamprosate (Campral) | 666 mg TID | 666 mg TID | Diarrhea | PO | ||||||||||
| Naltrexone (Vivitrol) |
|
| Injection site reactions, anxiety, syncope | PO, IM | ||||||||||
| Disulfiram | 125–500 mg/day | 250 mg/day | Bitter taste, impotence, drowsiness | PO | ||||||||||
| Nicotine Use Disorder | ||||||||||||||
| Bupropion, sustained-release (Zyban) | 150 mg daily or BID | 150 mg/day | Weight loss, constipation, agitation, xerostomia, nausea | PO | ||||||||||
| Nicotine |
|
| Oral irritation, headache, dyspepsia, nasal discomfort, cough, rhinitis | PO, intranasal, transdermal | ||||||||||
| Varenicline (Chantix)a | 1 mg BID up to 12 weeks | 0.5 mg/day | Nausea, abnormal dreams, headache | PO | ||||||||||
| ||||||||||||||
In addition to these approved medications, a variety of medications are used off-label for the management of several substance use disorders, including cocaine, methamphetamine, and cannabis.
Patient E, 54 years of age, presents with active opioid use disorder. He began using prescription opioids for back pain after having served in the infantry. His usage eventually resulted in his leaving military service prior to retirement, and he currently works in construction. The patient reports having relied on prescription opioids until three years ago, when he started using heroin. He buys 10 bundles per day of what is described as heroin but is most likely synthetic fentanyl. He has accidentally overdosed on two occasions, and emergency medical services used naloxone nasal spray to revive him. He is seeking care at an intensive outpatient program for treatment.
After reviewing goals of care with the patient, the clinician identifies that he has residual chronic back pain in addition to cravings for opioids when not using. The patient is diagnosed with opioid use disorder.
Patient E states that he recently sought care for his back pain and was referred to physical therapy and advised to take nonsteroidal anti-inflammatory drugs (NSAIDs) as needed for pain management. The possibility of methadone maintenance is discussed, but the patient does not want to have to go daily for dosing and is afraid that it will impair his ability to work around heavy machinery. Safety issues are also reviewed, and nasal naloxone is prescribed for emergency use. The patient's partner receives education on use as well.
The patient states he does not wish to take a medication, but he starts attending daily 12-step meetings and working with a drug counselor. Within five days, Patient E is able to successfully complete detoxification. However, after about six weeks he experiences a relapse. His partner administers nasal naloxone after finding him unconscious.
When Patient E returns for follow-up, various options for medication-assisted therapy are discussed, and he decides to enroll in buprenorphine/naloxone therapy, with a goal of eventually being maintained on monthly buprenorphine injection. The clinician emphasizes the need for continued counseling and the need to understand that, like all chronic diseases, patients with opioid use disorder are subject to relapse. The patient is advised of the importance of seeking help early in the event of a relapse. The biologic basis of substance use disorder is stressed; there is no judgement or shame.
Patient E does well on buprenorphine/naloxone therapy and is attending physical therapy for his back pain. He has continued with regular 12-step meetings and individual psychotherapy. At each follow-up appointment, the need to keep naloxone nasal spray available for emergency use is emphasized.
In the management of patients with mental health disorders, pharmacotherapy remains a vital part of optimal treatment. Only a portion of the psychopharmacologic options available have been discussed in this course. When providing care to special populations, such as pediatric patients and patients who are or may become pregnant, clinicians should consult the latest literature.
1. 1. Ozarin L. Diseases of the Mind: Highlights of American Psychiatry through 1900. Available at https://www.nlm.nih.gov/hmd/diseases/index.html. Last accessed September 17, 2024.
2. Szasz T. Coercion as Cure: A Critical History of Psychiatry. 2nd ed. New Brunswick, NJ: Transaction Publishers; 2010.
4. Ruffalo M. A Brief History of Electroconvulsive Therapy. Available at https://www.psychologytoday.com/us/blog/freud-fluoxetine/201811/brief-history-electroconvulsive-therapy. Last accessed September 17, 2024.
5. Ban TA. Fifty years chlorpromazine: a historical perspective. Neuropsychiatr Dis Treat. 2007;3(4):495-500.
7. Crilly J. The history of clozapine and its emergence in the US market: a review and analysis. Hist Psychiatry. 2007;18(1):39-60.
8. Atkin K, Kendall F, Gould D, et al. Neutropenia and agranulocytosis in patients receiving clozapine in the UK and Ireland. Br J Psychiatry. 1996;169:483-488.
9. Substance Abuse and Mental Health Services Administration. Key Substance Use and Mental Health Indicators in the United States: Results from the 2023 National Survey on Drug Use and Health. Available at https://www.samhsa.gov/data/report/2023-nsduh-annual-national-report. Last accessed September 16, 2024.
10. National Institute on Alcohol Abuse and Alcoholism. Medications Development Program. Available at https://www.niaaa.nih.gov/medications-development-program. Last accessed September 16, 2024.
11. American Society of Addiction Medicine. Public Policy Statement: Definition of Addiction. Available at https://www.asam.org/docs/default-source/public-policy-statements/1definition_of_addiction_long_4-11.pdf. Last accessed September 16, 2024.
13. Yaksh T, Wallace M. Opioids, analgesia, and pain management. In: Brunton L, Hilal-Dandan R, Knollmann BC (eds). Goodman and Gilman's The Pharmacological Basis of Therapeutics. 13th ed. New York, NY: McGraw-Hill; 2017: 355-386.
14. Havens JR, Walker R, Leukefeld CG. Prevalence of opioid analgesic injection among rural nonmedical opioid analgesic users. Drug Alcohol Depend. 2007;87(1):98-102.
15. Dydyk AM, Jain NK, Gupta M. Opioid Use Disorder. Treasure Island, FL: StatPearls Publishing; 2024.
16. Centers for Disease Control and Prevention. Health Alert: Increase in Fatal Drug Overdoses Across the United States Driven by Synthetic Opioids Before and During the COVID-19 Pandemic. Available at https://archive.cdc.gov/#/details?url=https://emergency.cdc.gov/han/2020/han00438.asp. Last accessed September 16, 2024.
17. Tamminga C. Antipsychotic Drugs. Available at https://www.merckmanuals.com/professional/psychiatric-disorders/schizophrenia-and-related-disorders/antipsychotic-drugs. Last accessed September 17, 2024.
18. Medscape. Drugs and Diseases. Available at https://reference.medscape.com. Last accessed September 17, 2024.
19. LexiComp. Available at https://online.lexi.com. Last accessed September 16, 2024.
20. Missouri Department of Mental Health. Abnormal Involuntary Movement Scale (AIMS). Available at https://dmh.mo.gov/media/21821/download. Last accessed September 16, 2024.
21. Reynolds GP, Kirk SL. Metabolic side effects of antipsychotic drug treatment—pharmacological mechanisms. Pharmacol Ther. 2010;125(1):169-179.
22. Roberts R, Neasham A, Lambrinudi C, Khan A. A quantitative analysis of antipsychotic prescribing trends for the treatment of schizophrenia in England and Wales. JRSM Open. 2018;9(4):2054270418758570.
23. U.S. Food and Drug Administration. Clozapine and the Risk of Neutropenia: A Guide for Healthcare Providers. Available at https://www.accessdata.fda.gov/drugsatfda_docs/rems/Clozapine_2021_02_18_Clozapine_and_the_Risk_of_Neutropenia_A_Guide_for_Healthcare_Providers.pdf. Last accessed September 16, 2024.
24. Vasavan Nair NP, Suranyi-Cadotte B, Schwartz G, et al. A clinical trial comparing intramuscular haloperidol decanoate and oral haloperidol in chronic schizophrenic patients: efficacy, safety, and dosage equivalence. J Clin Psychopharmacol. 1986;6(1 Suppl):30S-37S.
25. Zhornitsky S, Stip E. Oral versus long-acting injectable antipsychotics in the treatment of schizophrenia and special populations at risk for treatment nonadherence: a systematic review. Schizophr Res Treatment. 2012;2012:407171.
26. Marcinkowska M, Śniecikowska J, Fajkis N, Paśko P, Franczyk W, Kołaczkowski M. Management of dementia-related psychosis, agitation and aggression: a review of the pharmacology and clinical effects of potential drug candidates. CNS Drugs. 2020;34(3):243-268.
27. Reus VI, Fochtmann LJ, Eyler AE, et al. The American Psychiatric Association practice guideline on the use of antipsychotics to treat agitation or psychosis in patients with dementia. Am J Psychiatry. 2016;173(5):543-546.
28. Centers for Medicare & Medicaid Services. Atypical Antipsychotic Medications: Use in Pediatric Patients. Available at https://www.cms.gov/Medicare-Medicaid-Coordination/Fraud-Prevention/Medicaid-Integrity-Education/Pharmacy-Education-Materials/Downloads/atyp-antipsych-pediatric-factsheet11-14.pdf. Last accessed September 16, 2024.
29. Stübner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry. 2004;37(Suppl 1):S54-S64.
30. Berman BD. Neuroleptic malignant syndrome: a review for neurohospitalists. Neurohospitalist. 2011;1(1):41-47.
31. Brasic JR. Tardive Dyskinesia. Available at https://emedicine.medscape.com/article/1151826-overview. Last accessed September 17, 2021.
32. Ogino S, Miyamoto S, Miyake N, Yamaguchi N. Benefits and limits of anticholinergic use in schizophrenia: focusing on its effect on cognitive function. Psychiatry Clin Neurosci. 2014;68(1):37-49.
33. Ricciardi L, Pringsheim T, Barnes TRE, et al. Treatment recommendations for tardive dyskinesia. Can J Psychiatry. 2019;64(6):388-399.
34. National Institute of Mental Health. Statistics. Available at https://www.nimh.nih.gov/health/statistics/index.shtml. Last accessed September 17, 2024.
35. Gartlehner G, Hansen RA, Morgan LC, et al. Second-Generation Antidepressants in the Pharmacologic Treatment of Adult Depression. An Update of the 2007 Comparative Effectiveness Review. Comparative Effectiveness Reviews, No. 46. Rockville, MD: Agency for Healthcare Research and Quality; 2011.
36. Wang PS, Lane M, Olfson M, Pincus HA, Wells KB, Kessler RC. Twelve-month use of mental health services in the United States: results from the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):629-640.
37. Hasselmann HW. Ketamine as antidepressant? Current state and future perspectives. Curr Neuropharmacol. 2014;12(1):57-70.
38. Rosenblat JD, McIntyre RS, Alves GS, Fountoulakis KN, Carvalho AF. Beyond monoamines-novel targets for treatment-resistant depression: a comprehensive review. Curr Neuropharmacol. 2015;13(5):636-655.
39. Nasrallah HA. 10 recent paradigm shifts in the neurobiology and treatment of depression. Curr Psych. 2015;14(2).
40. Agency for Healthcare Research and Quality. Healthcare Horizon Scanning System Potential High-Impact Interventions: Priority Area 05: Depression and Other Mental Health Disorders. Rockville, MD: Agency for Healthcare Research and Quality; 2015.
41. Niciu MJ, Ionescu DF, Richards EM, Zarate CA Jr. Glutamate and its receptors in the pathophysiology and treatment of major depressive disorder. J Neural Transm (Vienna). 2014;121(8):907-924.
42. O'Connor AM, Bennett C, Stacey D, et al. Do patient decision aids meet effectiveness criteria of the international patient decision aid standards collaboration? A systematic review and meta-analysis. Med Decis Making. 2007;27(5):554-574.
43. American Psychiatric Association. Practice Guideline for the Treatment of Patients with Major Depressive Disorder. 3rd ed. Washington, DC: American Psychiatric Publishing, Inc.; 2010. (Reaffirmed October 31, 2015.)
44. American Psychological Association. Clinical Practice Guideline for the Treatment of Depression Across Three Age Cohorts. Available at https://www.apa.org/depression-guideline/guideline.pdf. Last accessed September 17, 2024.
45. Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry. 2001;62(Suppl 6):22-29.
46. Bostwick JM. A generalist's guide to treating patients with depression with an emphasis on using side effects to tailor antidepressant therapy. Mayo Clin Proc. 2010;85(6):538-550.
47. U.S. Department of Veterans Affairs, Department of Defense. VA/DoD Clinical Practice Guideline for the Management of Major Depressive Disorder. Washington, DC: U.S. Department of Veterans Affairs; 2016.
48. Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol. 2007;151(6):737-748.
49. Sinclair LI, Christmas DM, Hood SD, et al. Antidepressant-induced jitteriness/anxiety syndrome: systematic review. Br J Psychiatry. 2009;194(6):483-490.
50. Amsterdam JD. Monoamine oxidase inhibitor therapy in severe and resistant depression. Psychiatric Ann. 2006;36(9):607-613.
51. Fowler JS, Logan J, Azzaro AJ, et al. Reversible inhibitors of monoamine oxidase-A (RIMAs): robust, reversible inhibition of human brain MAO-A by CX157. Neuropsychopharmacology. 2010;35(3):623-631.
52. U.S. Food and Drug Administration. FDA Approves Emsam (Selegiline) as First Drug Patch for Depression [Archive]. Available at https://wayback.archive-it.org/7993/20170113023146/http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2006/ucm108607.htm. Last accessed September 17, 2024.
53. Caraci F, Leggio GM, Salomone S, Drago F. New drugs in psychiatry: focus on new pharmacological targets. F1000Res. 2017;6:397.
54. Deardorff WJ, Grossberg GT. A review of the clinical efficacy, safety and tolerability of the antidepressants vilazodone, levomilnacipran and vortioxetine. Expert Opin Pharmacother. 2014;15(17):2525-2542.
55. Sansone RA, Sansone LA. Emergency Psychiatry Update. Available at https://www.reliasmedia.com/articles/134475-emergency-psychiatry-update. Last accessed September 17, 2024.
56. Hu XH, Bull SA, Hunkeler EM, et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry. 2004;65(7):959-965.
57. Rizvi SJ, Kennedy SH. Management strategies for SSRI-induced sexual dysfunction. J Psychiatry Neurosci. 2013;38(5):E27-E28.
59. American Urological Association. Clinical Guidelines: Erectile Dysfunction: AUA Guideline (2018). Available at https://www.auanet.org/guidelines-and-quality/guidelines/erectile-dysfunction-(ed)-guideline. Last accessed September 17, 2024.
60. Möller HJ, Baldwin DS, Goodwin G, et al. Do SSRIs or antidepressants in general increase suicidality? WPA section on pharmacopsychiatry: consensus statement. Eur Arch Psychiatry Clin Neurosci. 2008;258(Suppl 3):3-23.
61. Leon AC, Solomon DA, Li C, et al. Antidepressants and risks of suicide and suicide attempts: a 27-year observational study. J Clin Psychiatry. 2011;72(5):580-586.
62. Kennedy SH, Lam RW, McIntyre RS, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder: section 3. pharmacological treatments. Can J Psychiatry. 2016;61(9):540-560.
63. Narayan V, Haddad PM. Antidepressant discontinuation manic states: a critical review of the literature and suggested diagnostic criteria. J Psychopharmacol. 2011;25(3):306-313.
64. U.K. Medicines and Healthcare Products Regulatory Agency. Selective Serotonin Reuptake Inhibitors (SSRIS) and Serotonin and Noradrenaline Reuptake Inhibitors (SNRIS): Use and Safety. Available at https://www.gov.uk/government/publications/ssris-and-snris-use-and-safety. Last accessed September 17, 2024.
65. Taylor D, Stewart S, Connolly A. Antidepressant withdrawal symptoms: telephone calls to a national medication helpline. J Affect Disord. 2006;95(1):129-133.
66. Fava GA, Gatti A, Belaise C, Guidi J, Offidani E. Withdrawal symptoms after selective serotonin reuptake inhibitor discontinuation: a systematic review. Psychother Psychosom. 2015;84(2):72-81.
67. Renoir T. Selective serotonin reuptake inhibitor antidepressant treatment discontinuation syndrome: a review of the clinical evidence and the possible mechanisms involved. Front Pharmacol. 2013;4:45.
68. Michelson D, Fava M, Amsterdam J, et al. Interruption of selective serotonin reuptake inhibitor treatment: double-blind, placebo-controlled trial. Br J Psychiatry. 2000;176:363-368.
69. Tint A, Haddad PM, Anderson IM. The effect of rate of antidepressant tapering on the incidence of discontinuation symptoms: a randomized study. J Psychopharmacol. 2008;22(3):330-332.
70. National Institute for Health and Clinical Excellence. Depression: The Treatment and Management of Depression in Adults. London: The British Psychological Society and the Royal College of Psychiatrists; 2010.
71. U.S. Food and Drug Administration. Suicidality in Children and Adolescents Being Treated With Antidepressant Medications. Available at https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/suicidality-children-and-adolescents-being-treated-antidepressant-medications. Last accessed September 17, 2024.
72. Garland EJ, Kutcher S, Virani A, Elbe D. Update on the use of SSRIs and SNRIs with children and adolescents in clinical practice.J Can Acad Child Adolesc Psychiatry. 2016;25(1):4-10.
73. Ménard C, Hodes GE, Russo SJ. Pathogenesis of depression: insights from human and rodent studies. Neuroscience. 2016;321:138-162.
74. Mathews DC, Henter ID, Zarate CA. Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs. 2012;72(10):1313-1333.
75. Kaster MP, Moretti M, Cunha MP, Rodrigues AL. Novel approaches for the management of depressive disorders. Eur J Pharmacol. 2016;771:236-240.
76. DeWilde KE, Levitch CF, Murrough JW, Mathew SJ, Iosifescu DV. The promise of ketamine for treatment-resistant depression: current evidence and future directions. Ann N Y Acad Sci. 2015;1345:47-58.
77. McIntyre RS, Filteau MJ, Martin L, et al. Treatment-resistant depression: definitions, review of the evidence, and algorithmic approach. J Affect Disord. 2014;156:1-7.
78. Kennedy SH. A review of antidepressant therapy in primary care: current practices and future directions. Prim Care Companion CNS Disord. 2013;15(2):12r01420.
79. Connolly KR, Thase ME. The clinical management of bipolar disorder: a review of evidence-based guidelines. Prim Care Companion CNS Disord. 2011;13(4):10r01097.
80. Butler M, Urosevic S, Desai P, et al. Treatment for Bipolar Disorder in Adults: A Systematic Review. Rockville, MD: Agency for Healthcare Research and Quality; 2018.
81. McClintock SM, Reti IM, Carpenter LL, et al. Consensus recommendations for the clinical application of repetitive transcranial magnetic stimulation (rTMS) in the treatment of depression. J Clin Psychiatry. 2018;79(1):16cs10905.
82. Perrin JS, Merz S, Bennett DM, et al. Electroconvulsive therapy reduces frontal cortical connectivity in severe depressive disorder.Proc Natl Acad Sci U S A. 2012;109(14):5464-5468.
83. Milev RV, Giacobbe P, Kennedy SH, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 clinical guidelines for the management of adults with major depressive disorder: section 4. neurostimulation treatments. Can J Psychiatry. 2016;61(9):561-575.
84. Malhi GS, Bassett D, Boyce P, et al. Royal Australian and New Zealand College of Psychiatrists clinical practice guidelines for mood disorders. Aust N Z J Psychiatry. 2015;49(12):1087-1206.
85. Brunoni AR, Boggio PS, De Raedt R, et al. Cognitive control therapy and transcranial direct current stimulation for depression: a randomized, double-blinded, controlled trial. J Affect Disord. 2014;162:43-49.
86. Garay RP, Zarate CA Jr, Charpeaud T, et al. Investigational drugs in recent clinical trials for treatment-resistant depression. Expert Rev Neurother. 2017;17(6):593-609.
87. Jaso BA, Niciu MJ, Iadarola ND, et al. Therapeutic modulation of glutamate receptors in major depressive disorder. Curr Neuropharmacol. 2017;15(1):57-70.
88. Kishimoto T, Chawla JM, Hagi K, et al. Single-dose infusion ketamine and non-ketamine N-methyl-D-aspartate receptor antagonists for unipolar and bipolar depression: a meta-analysis of efficacy, safety and time trajectories. Psychol Med. 2016;46(7):1459-1472.
89. U.S. Food and Drug Administration. FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression; Available Only at Certified Doctor's Office or Clinic. Available at https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified. Last accessed September 17, 2024.
90. Shultz E, Malone DA Jr. A practical approach to prescribing antidepressants. Cleve Clin J Med. 2013;80(10):625-631.
91. Kumar Singh L, Haque Nizamie S, Sayeed A, Kumar Praharaj S. Improving tolerability of lithium with a once-daily dosing schedule. Am J Ther. 2011;18(4):288-291.
92. Munk-Olsen T, Liu X, Viktorin A, et al. Maternal and infant outcomes associated with lithium use in pregnancy: an international collaborative meta-analysis of six cohort studies. Lancet Psychiatry. 2018;5(8):644-652.
93. Goren JL, Levin GM. Mania with bupropion: a dose-related phenomenon? Ann Pharmacother. 2000;34(5):619-621.
94. National Institute of Mental Health. Any Anxiety Disorder. Available at https://www.nimh.nih.gov/health/statistics/any-anxiety-disorder. Last accessed September 17, 2024.
95. National Institute of Mental Health. Anxiety Disorders. Available at https://www.nimh.nih.gov/health/topics/anxiety-disorders/index.shtml. Last accessed September 17, 2024.
96. Grassi M, Caldirola D, Di Chiaro NV, et al. Are respiratory abnormalities specific for panic disorder? A meta-analysis. Neuropsychobiology. 2014;70(1):52-60.
98. Klein DF, Fink M. Psychiatric reaction patterns to imipramine. Am J Psychiatry. 1962;119:432-438.
99. Dell'osso B, Lader M. Do benzodiazepines still deserve a major role in the treatment of psychiatric disorders? A critical reappraisal. Eur Psychiatry. 2013;28(1):7-20.
100. Spiegel DA. Efficacy studies of alprazolam in panic disorder. Psychopharmacol Bull. 1998;34(2):191-195.
101. American Psychiatric Association. Practice Guideline for the Treatment of Patients with Panic Disorder. 2nd ed. Washington, DC: American Psychiatric Publishing; 2009.
102. Sayed S, Horn SR, Murrough JW. Current treatment for anxiety and obsessive-compulsive disorders. Curr Treat Options Psychiatry. 2014;1:248-262.
103. Locke AB, Kirst N, Shultz CG. Diagnosis and management of generalized anxiety disorder and panic disorder in adults. Am Fam Physician. 2015;91(9):617-624.
104. Mavissakalian MR. Imipramine vs. sertraline in panic disorder: 24-week treatment completers. Ann Clin Psychiatry. 2003;15(3-4):171-180.
105. Schmitt R, Gazalle FK, Lima MS, Cunha A, Souza J, Kapczinski F. The efficacy of antidepressants for generalized anxiety disorder: a systematic review and meta-analysis. Rev Bras Psiquiatr. 2005;27(1):18-24.
106. Farach FJ, Pruitta LD, Jun JJ, Jerud AB, Zoellner LA, Roy-Byrne PP. Pharmacological treatment of anxiety disorders: current treatments and future directions. J Anxiety Disord. 2012;26(8):833-843.
107. Santarsieri D, Schwartz TL. Antidepressant efficacy and side-effect burden: a quick guide for clinicians. Drugs Context. 2015;4:212290.
108. Grohol JM, Wright SA. Top 25 Psychiatric Medication Prescriptions for 2020. Available at https://psychcentral.com/blog/top-25-psychiatric-medications-for-2020. Last accessed September 17, 2024.
109. Johnson B, Streltzer J. Risks associated with long-term benzodiazepine use. Am Fam Physician. 2013;88(4):224-226.
110. Royal Australian College of General Practitioners. Prescribing Drugs of Dependence in General Practice, Part B: Benzodiazepines. Available at https://www.racgp.org.au/clinical-resources/clinical-guidelines/key-racgp-guidelines/view-all-racgp-guidelines/drugs-of-dependence/part-b. Last accessed September 17, 2024.
111. Royal Australian and New Zealand College of Psychiatrists. Australian and New Zealand clinical practice guidelines for the treatment of panic disorder and agoraphobia. Aust N Z J Psychiatry. 2003;37(6):641-656.
112. Bandelow B, Reitt M, Röver C, Michaelis S, Görlich Y, Wedekind D. Efficacy of treatments for anxiety disorders: a meta-analysis. Int Clin Psychopharmacol. 2015;30(4):183-192.
114. Roy-Byrne P. Treatment-refractory anxiety; definition, risk factors, and treatment challenges. Dialogues Clin Neurosci. 2015;17(2):191-206.
115. Westra HA, Stewart SH, Conrad BE. Naturalistic manner of benzodiazepine use and cognitive behavioral therapy outcome in panic disorder with agoraphobia. J Anxiety Disord. 2002;16(3):233-246.
116. Watanabe N, Churchill R, Furukawa T. Combination of psychotherapy and benzodiazepines versus either therapy alone for panic disorder: a systematic review. BMC Psychiatry. 2007;7:18.
117. Goddard AW, Brouette T, Almai A, Jetty P, Woods SW, Charney D. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
119. Hood SD, Norman A, Hince DA, et al. Benzodiazepine dependence and its treatment with low dose flumazenil. Br J Clin Pharmacol. 2014;77(2):285-294.
120. Pollack MH, Van Ameringen M, Simon NM, et al. A double-blind randomized controlled trial of augmentation and switch strategies for refractory social anxiety disorder. Am J Psychiatry. 2014;171(1):44-53.
121. Nutt DJ. Overview of diagnosis and drug treatments of anxiety disorders. CNS Spectr. 2005;10(1):49-56.
122. Baldwin DS, Anderson IM, Nutt DJ, et al. Evidence-based pharmacological treatment of anxiety disorders, post-traumatic stress disorder and obsessive-compulsive disorder: a revision of the 2005 guidelines from the British Association for Psychopharmacology. J Psychopharmacol. 2014;28(5):403-439.
123. Pfizer Pharmaceuticals. Xanax (Alprazolam). Available at https://www.pfizer.com/products/product-detail/xanax. Last accessed September 17, 2024.
124. Ashton H. Risks of dependence on benzodiazepine drugs: a major problem of long-term treatment. BMJ. 1989;298(6666):103-104.
125. Stewart SA. The effects of benzodiazepines on cognition. J Clin Psychiatry. 2005;66(Suppl 2):9-13.
126. Movig KL, Mathjssen MP, Nagel PH, et al. Psychoactive substance use and the risk of motor vehicle accidents. Accid Anal Prev. 2004;36(4):631-636.
127. Finkle WD, Der JS, Greenland S, et al. Risk of fractures requiring hospitalization after an initial prescription for zolpidem, alprazolam, lorazepam, or diazepam in older adults. J Am Geriatr Soc. 2011;59(10):1883-1890.
128. Liebrenz M, Gehring M-T, Buadze A, Caflisch C. High-dose benzodiazepine dependence: a qualitative study of patients' perception on cessation and withdrawal. BMC Psychiatry. 2015;15:116.
129. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
130. U.S. Food and Drug Administration. FDA Updates and Press Announcements on Nitrosamine in Varenicline (Chantix). Available at https://www.fda.gov/drugs/drug-safety-and-availability/fda-updates-and-press-announcements-nitrosamine-varenicline-chantix. Last accessed September 5, 2024.
131. Fabre LF, Brown CS, Smith LC, Derogatis LR. Gepirone-ER treatment of hypoactive sexual desire disorder (HSDD) associated with depression in women. J Sex Med. 2011;8(5):1411-1419.
132. Fabre LF, Clayton AH, Smith LC, Goldstein I, Derogatis LR. The effect of gepirone-ER in the treatment of sexual dysfunction in depressed men. J Sex Med. 2012;9(3):821-829.
133. Fabre-Kramer. EXXUA for Major Depression. Available at https://fabrekramer.com/exxua-for-major-depression. Last accessed September 5, 2024.
1. American Psychiatric Association. Practice Guideline for the Treatment of Patients with Schizophrenia. 3rd ed. Washington, DC: American Psychiatric Association; 2020. Available at https://psychiatryonline.org/doi/full/10.1176/appi.books.9780890424841.Schizophrenia02. Last accessed September 27, 2024.
2. American Psychiatric Association. Practice Guideline for the Treatment of Patients with Major Depressive Disorder. 3rd ed. Arlington, VA: American Psychiatric Association; 2010. Available at https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. Last accessed September 27, 2024.
3. National Collaborating Centre for Mental Health, National Collaborating Centre for Primary Care. Bipolar Disorder: Assessment and Management. London: National Institute for Health and Clinical Excellence; 2023. Available at https://www.nice.org.uk/guidance/cg185. Last accessed September 27, 2024.
4. National Collaborating Centre for Mental Health, National Collaborating Centre for Primary Care. Generalised Anxiety Disorder and Panic Disorder (with or without Agoraphobia) in Adults: Management in Primary, Secondary and Community Care. London: National Institute for Health and Clinical Excellence; 2020. Available at https://www.nice.org.uk/guidance/cg113. Last accessed September 27, 2024.
5. Crotty K, Freedman KI, Kampman KM. Executive summary of the focused update of the ASAM national practice guideline for the treatment of opioid use disorder. Journal of Addiction Medicine. 2020;14(2):99-112. Available at https://www.asam.org/docs/default-source/quality-science/jam-exec-summary.pdf. Last accessed September 27, 2024.
Mention of commercial products does not indicate endorsement.