Techniques to ameliorate pain have been actively sought since the earliest recorded history of science and medicine. Although opioids are generally effective, the associated risks and side effects make their use undesirable in many patients. In an effort to decrease the use of opioids, it is vital for clinicians to first consider other agents to control pain. Combining various classes of drugs, in lower doses, can help control pain while decreasing side effects. This course will focus on the science behind multimodal pharmacologic pain management and its efficacy.
This course is designed for nurses involved in the care of patients with pain.
The purpose of this course is to provide healthcare providers with a clear understanding of the concept of multimodal pharmacotherapy for pain relief, including available classes of analgesics.
Upon completion of this course, you should be able to:
- Describe the underlying pathophysiology of pain.
- Outline the different types of pain.
- Discuss the mechanism of action and clinical use of opioids in the management of pain.
- Compare and contrast other analgesic agents that can be used in a multimodal approach to pain management, including nonsteroidal anti-inflammatory drugs (NSAIDs), antidepressants, and local anesthetics.
- Analyze approaches to multimodal pharmacotherapy for pain management.
Richard E. Haas, BSN, MSN, EdM, PhD, CRNA, LTC US Army Nurse Corps (Retired), is a retired nurse anesthetist and prehospital registered nurse (instructor) who has published extensively in various areas of healthcare research while providing clinical care in arenas ranging from academic medical centers to austere environments in the third world during both wartime and peacetime. He has a bachelor’s degree in nursing from Georgetown University, Master’s degrees in education (Boston University) and nursing specializing in anesthesia (State University of New York in Buffalo and U.S. Army), and a PhD from the University of South Carolina. He is a retired lieutenant colonel in the U.S. Army Nurse Corps. He has taught nursing anesthesia, pharmacology, and physiology; mentored students in doctoral programs; and used advanced patient simulation to train students. Dr. Haas has worked in clinical, administrative, education, and research roles. He continues to work as an independent consultant, while taking more time to enjoy life with his wife of nearly 50 years and their children and grandchildren.
Contributing faculty, Richard E. Haas, BSN, MSN, EdM, PhD, CRNA, LTC US Army Nurse Corps (Retired), has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Mary Franks, MSN, APRN, FNP-C
The division planner has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Sarah Campbell
The Director of Development and Academic Affairs has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
The purpose of NetCE is to provide challenging curricula to assist healthcare professionals to raise their levels of expertise while fulfilling their continuing education requirements, thereby improving the quality of healthcare.
Our contributing faculty members have taken care to ensure that the information and recommendations are accurate and compatible with the standards generally accepted at the time of publication. The publisher disclaims any liability, loss or damage incurred as a consequence, directly or indirectly, of the use and application of any of the contents. Participants are cautioned about the potential risk of using limited knowledge when integrating new techniques into practice.
It is the policy of NetCE not to accept commercial support. Furthermore, commercial interests are prohibited from distributing or providing access to this activity to learners.
Supported browsers for Windows include Microsoft Internet Explorer 9.0 and up, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Supported browsers for Macintosh include Safari, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Other operating systems and browsers that include complete implementations of ECMAScript edition 3 and CSS 2.0 may work, but are not supported. Supported browsers must utilize the TLS encryption protocol v1.1 or v1.2 in order to connect to pages that require a secured HTTPS connection. TLS v1.0 is not supported.
The role of implicit biases on healthcare outcomes has become a concern, as there is some evidence that implicit biases contribute to health disparities, professionals' attitudes toward and interactions with patients, quality of care, diagnoses, and treatment decisions. This may produce differences in help-seeking, diagnoses, and ultimately treatments and interventions. Implicit biases may also unwittingly produce professional behaviors, attitudes, and interactions that reduce patients' trust and comfort with their provider, leading to earlier termination of visits and/or reduced adherence and follow-up. Disadvantaged groups are marginalized in the healthcare system and vulnerable on multiple levels; health professionals' implicit biases can further exacerbate these existing disadvantages.
Interventions or strategies designed to reduce implicit bias may be categorized as change-based or control-based. Change-based interventions focus on reducing or changing cognitive associations underlying implicit biases. These interventions might include challenging stereotypes. Conversely, control-based interventions involve reducing the effects of the implicit bias on the individual's behaviors. These strategies include increasing awareness of biased thoughts and responses. The two types of interventions are not mutually exclusive and may be used synergistically.
#35271: Multimodal Pharmacotherapy for Pain Management
Pain is one of the most potent safeguards to homeostasis. Experiencing pain allows humans to learn which activities or substances might cause irreparable damage to the body, helping to avoid engaging in those activities or ingesting those substances. However, this system is not perfect. Techniques to ameliorate pain have been actively sought since the earliest recorded history of science and medicine. Opioids, alcohol, mandrakes, and cannabis have all been used through the years in an attempt to mitigate pain [1].
From the 1860s until the 1950s, morphine was the pre-eminent opioid used for the control of pain. It was easily produced, especially as the pharmacotherapeutic industry became increasingly more refined and developed. In the 1950s, researcher Paul Janssen began his work on the synthesis of a new opioid called fentanyl [2]. In 1960, fentanyl was synthesized, and its use became widespread as a result of its safety and efficacy. Two derivatives of fentanyl followed in the 1970s: alfentanil (with a more rapid offset) and sufentanil (with increased potency) [2]. Opioids continue to be the mainstay of severe pain relief.
Even as researchers have lauded the effects of opioids for pain relief, they decried the problems with the use of opioids, particularly the risks of abuse and addiction. In the decade following their widespread use in the Civil War, physicians began to refer to opioid use disorder as the "soldier's disease" [3]. Unfortunately, knowledge of the problem has not led to an immediate solution. Problems with opioid abuse, including use disorder and diversion for illicit use, continue. At the same time, opioids (including fentanyl and its congeners) are widely used for the control of severe pain. There is evidence that even appropriate use of opioids may lead to use disorder and diversion in a subset of patients [4].
In an effort to decrease the use of opioids, it is vital for clinicians to first consider other agents to control pain. Combining various classes of drugs, in lower doses, can help control pain while decreasing side effects. One large dose of an opioid may be effective, but the preferred approach may be to use less or no opioid and to combine other agents, including anti-inflammatories, local anesthetics, alpha-2 receptor agonists, and others, to attain pain relief without the risks associated with opioids. This course will focus on the science behind multimodal pharmacologic pain management and its efficacy.
Management of pain is a highly complex and patient-centered specialty. Clear understanding of the underlying anatomy and physiology is required to make the correct selection of pharmacologic interventions.
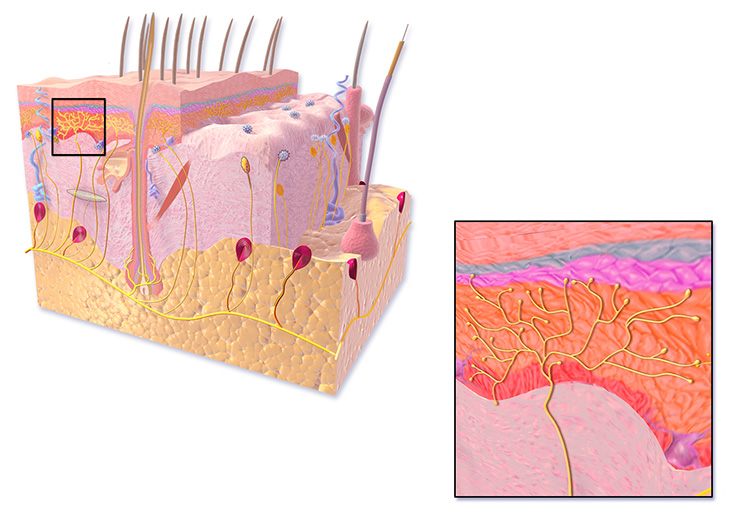
The human body has a number of specialized receptors in the skin, viscera, and periosteum of the bones that send impulses to the spinal cord and brain reporting the presence of pain (Table 1) [5]. Of particular interest to those treating pain is the free nerve ending (FNE) (Figure 1). There are several types of these unmyelinated nerve endings, and the ones most related to pain and tissue damage information processing are type IVa [6]. FNEs can send an impulse in the form of an action potential into the pain pathways as the result of tissue damage (secondary to trauma or heat) or tissue deformity (severe pressure resulting in tissue destruction). It is helpful to briefly review the concept of the action potential before moving on to an examination of the FNE.
TYPES OF SENSORY RECEPTORS
| Type | Function | Location |
|---|---|---|
| Free nerve ending | Transmit pain and temperature | Skin, periosteum, arterial walls, joint surfaces |
| Pacinian (lamellar) corpuscle | Pressure | Skin |
| Meissner (tactile) corpuscle | Touch | Skin |
| Muscle spindle | Stretch and pressure | Skeletal muscle |
| Golgi tendon apparatus | Stretch and pressure | Tendons |
| Kinesthetic receptor | Three-dimensional location (proprioception) | Joints |
Action potentials are changes in polarity along a nerve based on ion flow into and out of the nerve cell. As the action potential travels down the length of the cell, it will end at a point in the nervous system that results in some form of output, either physical (muscle movement) or experiential (pain).Figure 2 shows some of the details of an action potential and provides an extended explanation of their formation. Of particular importance is the quantity of ions moving at any specific time. The primary ions moving after stimulation and reaching threshold are Na+ (sodium, into the cell), K+ (potassium, out of the cell), and Cl- (chloride, into the cell). After the nerve has fired, the sodium/potassium adenosine triphosphate (ATP)-ase pump works to move sodium out of the cell and potassium back into the cell. A pump is needed because the ions are moving against their gradients, and energy is required in the form of ATP to power the pump [7].
ION MOVEMENT IN ACTION POTENTIAL FORMATION
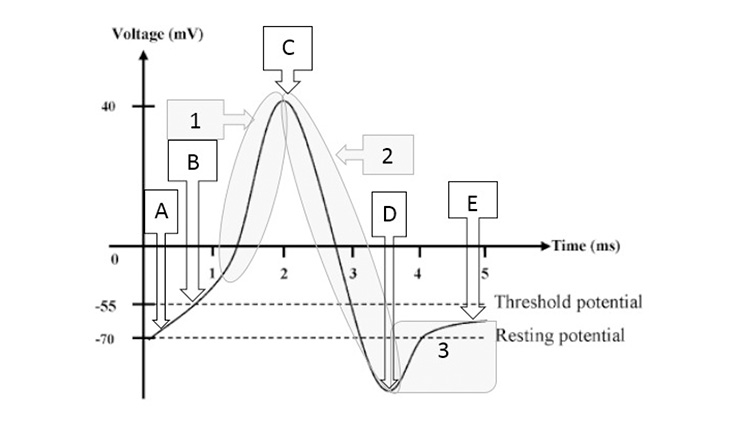 |
| Point A above shows the resting membrane potential of the nerve. At rest, a certain degree of leakage of both the sodium (Na+) into the cell and potassium (K+) out of the cell occurs. When a painful stimulus is experienced in the periphery, impulses will arrive at some point along a secondary nerve. Note the gradual upslope of the potential within the cell (the section between points A and B). In this example, painful impulses are coming into a section of nerve tissue from a free nerve ending, resulting in opening voltage-gated Na+ channels, resulting in an influx of Na+ that is faster than the outward leakage of K+, leading to an increasing membrane potential. Once membrane potential reaches threshold (point B), a large number of voltage-gated Na+ channels open, resulting in a large shift of Na+ ions from the extracellular fluid to the intracellular fluid, causing the initiation of an action potential (shaded area 1). Voltage-gated K+ and chloride (Cl-) channels then open, allowing K+ efflux and Cl- influx, dropping membrane potential (shaded area 2). The movement of K+ and Cl- is so great that an overshoot occurs, and the nerve hyperpolarizes (or falls below resting membrane potential, point D and shaded area 3). The Na+/K+ ATP-ase pump begins to work (shaded area 3) and equalizes Na+ and K+ gradients to result in a return to normal resting membrane potential. |
A variable but set amount of nervous action potential impulses is required to reach threshold. The nerve must receive a sufficient number of input signals (or stimuli) to move its membrane potential above threshold and fire an impulse, sending, in this case, a message of pain along a nerve pathway. Understanding the science behind the action potential and its formation is crucial for the healthcare provider, as many analgesic drugs work by altering the transmissibility of signals sent along the nerve. If the action potential formed in the FNE can be blocked or altered, painful sensations can be mitigated or eliminated.
The FNEs are one of the sets of tissue receptors that can be stimulated to send impulses (action potentials) along the length of the nerve for further interpretation either in the spinal cord or various sections of the brain. Despite the study of FNEs over the past decade, the exact mechanism of action is still unknown. There are several postulations about how stimuli cause the firing of FNEs, initiating action potentials and resulting in the experience of pain. FNEs are also referred to as nociceptors. While nociceptors are often described as pain receptors, the purpose of the nociceptor is to alert the body to the presence of tissue damage [8]. FNEs are small structures, with a thin layer of Schwann cells surrounding them. They are branched in appearance and have small varicosities containing mitochondria, vesicles, and an axonal reticulum (analogous to the sarcoplasmic reticulum or endoplasmic reticulum in other cell types) [5,9]. The vesicles contain numerous neuropeptides, including substance P and calcitonin gene-related peptide (CGRP), both of which are crucial in spreading action potentials during conditions of tissue destruction resulting in pain [9]. Figure 2 illustrates varicosities in the distal ends of the FNE, but these structures are present throughout the FNEs until they reach larger nerves.
There are two primary types of nerves that carry pain data elicited by stimulation of an FNE: type Aδ or myelinated (fast) nerve fibers and type C or unmyelinated (slow) nerve fibers [5,8]. Myelinated fibers are insulated with Schwann cells, but with gaps (nodes of Ranvier) in which the nerve fiber is exposed to the environment of the extracellular fluid. The myelinated fibers are also referred to as fast fibers because the action potentials can skip between the nodes of Ranvier in a process called saltatory conduction (rather than traveling the entire length of the axon). Sharp or acute pain, especially from traumatic injury, is usually processed in this fashion.
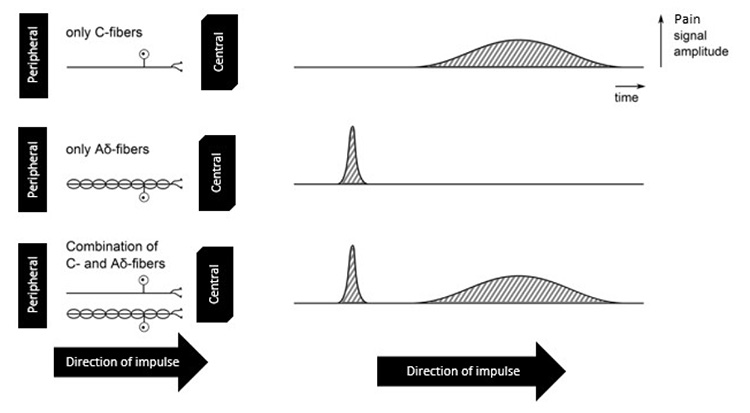
Type C fibers move impulses more slowly. While myelinated fibers can carry action potential impulses at speeds of 5–30 meters per second, unmyelinated fibers have speeds of 0.4–1.4 meters per second [8]. Most individuals have experienced this type of pain differential speed in their own lives: an acute accidental injury such as cutting one's finger while cooking results in an immediate response followed by an aching sensation. This is because both type A and type C nerve fibers travel in bundles together, and the stimulation of one nerve makes the stimulation of a nearby nerve easier. An example of this phenomenon is seen in Figure 3. The solid line represents an unmyelinated nerve (type C), while the line with nodes represents a myelinated nerve (type Aδ). If the nerves travel together from the site of injury, the patient experiences two waves of pain. Myelinated fibers carrying information about pain tend to be highly localized, dependent on the density of FNEs in the area of injury. Unmyelinated fibers tend to carry information related to aching or less acute or localized pain. A significant amount of visceral pain tends to be carried by unmyelinated fibers, making diagnoses of this type of pain difficult.
Pain pathways are complex routes over which action potentials are sent from the peripheral nerves to the central nervous system (CNS). An interruption of the pathway at any point tends to mitigate the degree of pain felt by the patient and, in some cases, may alleviate pain entirely. This can be accomplished by blocking some part of the pathway with a local anesthetic or by administering an agent(s) that drives the resting membrane potentials of neurons in a more negative fashion or interrupts the pathway within the brain. One of the key aspects of multimodal pharmacologic pain relief is to use smaller doses of various agents that work at different points along the pathway, minimizing adverse side effects while maximizing the number of sites of action.
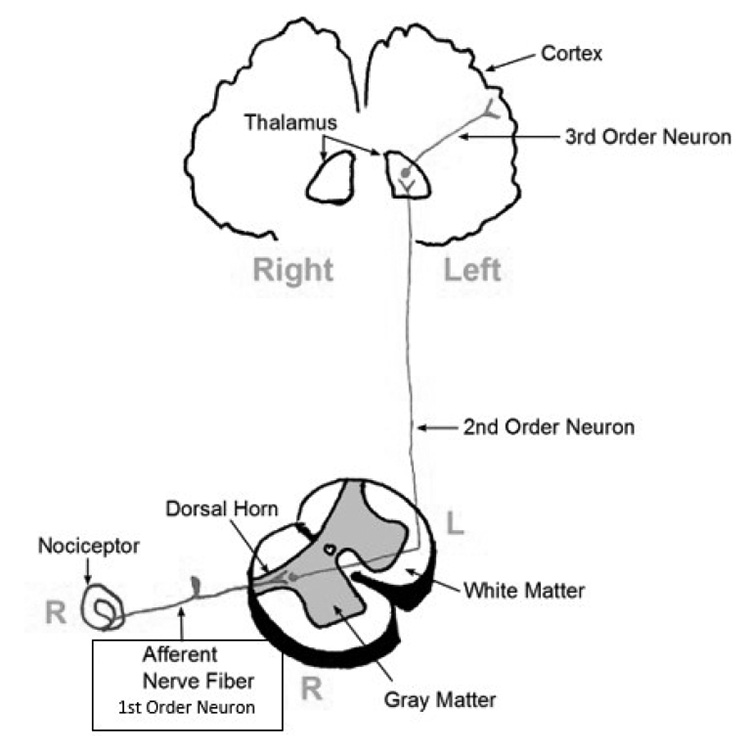
Pain and other impulses originate in the peripheral nervous system (PNS), enter the dorsal horn of the vertebra, and then ascend to the brain along the spinothalamic tract. This is a three-neuron pathway containing first-, second-, and third-order neurons. In Figure 4, the spinothalamic tract can be traced from the primary afferent nerve (receiving the pain signals at the site of injury) to the spinal cord, entering via the dorsal root of the cord. At this point, the first-order neuron synapses with a second-order neuron. Upon entry into the cord, the second-order neuron crosses from the right to the left (or left to right, if it enters the left dorsal root). This is referred to as decussation. The second-order neuron then rises up the cord in either the anterior or later spinothalamic tract, synapsing with a third-order neuron in the thalamus. This neuron leads to the sensory cortex in the brain, which in turn interprets the exact location and degree of pain.
Because pain is adaptive in nature, it is important that signals are sent to the central nervous system to ensure the body responds to maintain homeostasis. As discussed, FNEs exposed to noxious stimuli send action potentials down a specific pain receptor pathway to insure its arrival in the central nervous system [9,10,11,12]. Recall also that action potential generation is dependent upon the release of neurotransmitters, which in turn raise the nerve's membrane potential above threshold. While many neurotransmitters do this, a significant number function to lower resting membrane potential. This environment, in the presence of injury, has been referred to as "inflammatory soup," as a representation of the numerous and diverse substances involved in responses to pain [7]. Table 2 provides a synopsis of most of these substances, focusing on those with the greatest impact in pain management. For nearly every substance, there is some form of pharmacologic antagonist available or in development to nullify its excitatory effect [5,10,11,12,13,14,15]. While Table 2 provides a brief glimpse at how neurons can be excited by local peptides and neurotransmitters, the actual mechanisms by which these changes are made are incredibly complex.
SUBSTANCES AFFECTING THE TRANSMISSION OF IMPULSES IN FREE NERVE ENDINGS AND SOMATIC NERVES
| Substance | Description |
|---|---|
| Bradykinin | Bradykinin is a vasodilator that increases capillary permeability, increases migration of white blood cells, and increases free radicals in inflamed tissue and significantly excites pain receptors. |
| Calcitonin gene-related peptide (CGRP) | Stimulation of the free nerve endings results in the release of CGRP from the neuron, sensitizing it to stimuli and making the neuron hyperactive. |
| Norepinephrine | Pain stimulates the sympathetic nervous system, leading to the release of norepinephrine, which has an excitatory effect on the neuron. |
| Glutamate | Glutamate is an endogenous and highly excitatory neurotransmitter that binds at both the NMDA and AMPA receptors to excite the neuron and facilitate pain transmission. |
| Histamine | A ubiquitous substance throughout the body, histamine is released by mast cells and binds with excitatory receptors on the neurons and other cells. |
| Tachykinin | Tachykinins are a broad family of neuropeptides, including substance P, neurokinin A, and neurokinin B, released in response to pain or inflammation. They bind with neurokinin receptors, resulting in increasing excitatory stimulation of the neuron. |
| Serotonin (5-HT) | During inflammation, 5-HT is released from platelets in the area of injury. In turn, these bind with 5-HT2A and 5-HT3 receptors, resulting in excitation of the nerve. |
| Prostaglandin | One of the most crucial substances in pain management, prostaglandin sensitizes all aspects of excitatory phenomena in neurons. They are produced from the cell's arachidonic acid supply via the cyclo-oxygenase and lipoxygenase pathways. |
| Cytokine | Cytokines increase synaptic excitatory transmission in neurons and are represented by such substances as TNF and interleukins (e.g., IL-1b, IL-6). |
| AMPA = α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, NMDA = N-methyl-D-aspartate, TNF = tumor necrosis factor. | |
There is a large number of receptors on the neuron's cell membrane, most of which can bind with a specific molecular neurotransmitter. Nearly every drug administered in multimodal pain therapy interacts with one or more of these receptors.
Figure 5 is a complex diagram of the ascending pain pathways. Starting in the lower left corner (with the stimulus), the pain pathway can be seen tracking along both Ad and C fibers and entering the dorsal root of the spinal cord. The peripheral nerve synapses with a second-order nerve (Box 1) that decussates across the spinal cord; activating or inhibiting the interneurons in the lamina of the dorsal root can stop the impulse from propagating. The pain pathway then begins to ascend the anterior and lateral spinothalamic tracts toward the brain (Box 2). In the neck area of the diagram, the spinothalamic tract has been highlighted (Box 3), and it continues to ascend into the thalamus in the brain. The second-order neurons synapse in a special area of the thalamus called the ventrobasal complex (Box 4) [5]. The thalamus, when activated, is thought to cause the conscious perception of pain and provides an anatomic location for the cell bodies of the third-order neurons [5]. The third-order neurons then ascend to the somatosensory cortex, allowing the patient to localize and quantify the painful stimulus (Box 5).
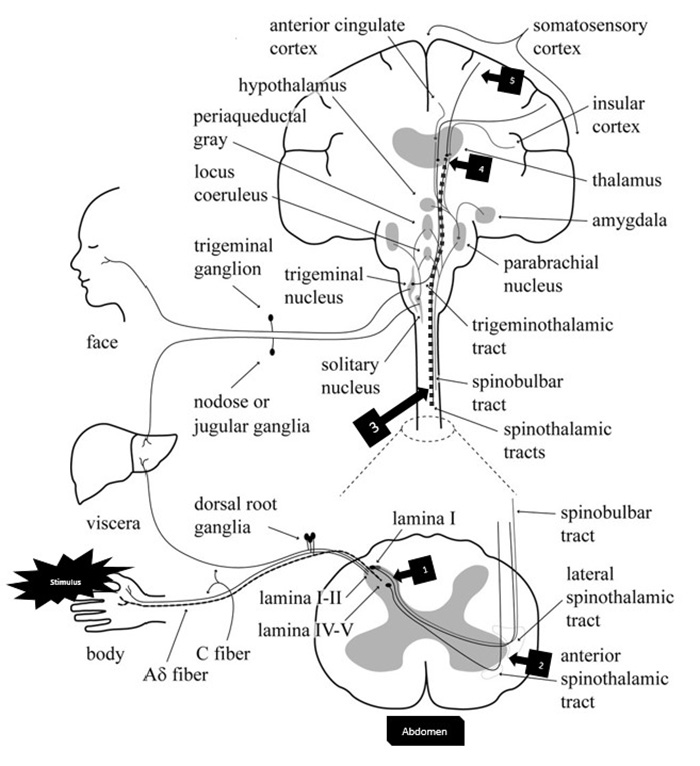
The thalamus also has neuronal branches that help to stimulate the reticular activating system, the portion of the brain responsible for sleep and waking [5]. The thalamus has numerous projections into other areas of the brain, including the prefrontal cortex and the amygdala, the latter of which is part of the limbic system [16,17]. The projections of the neurons into the limbic system account for the suffering aspect of pain, where the sensation is overlaid with an emotional experience. As pain is important in preventing homeostasis damage, including an emotional response to pain (in addition to a sensory response) helps ensure the person experiencing pain will avoid the stimulus that led to the pain.
The hippocampus is another area that receives neuronal impulses during painful stimuli [17]. Previous studies have linked decreases in hippocampal volume to major depression; however, the hippocampus also helps process and modify nociceptive stimulation [18,19]. Further, the hippocampus is the primary site for implanting memories [17].
Patients who are exposed to chronic pain or chronic stress may develop severe pain syndromes refractory to usual treatment. These pain syndromes have been associated with increased production of tumor necrosis factor-alpha (TNFα), a proinflammatory cytokine that sensitizes the patient to increased levels of pain secondary to local inflammation [19]. These inflammatory processes also atrophy the hippocampus, which has been associated with major depressive disorder [20]. It is not unusual, therefore, for unremitted or inadequately treated pain to co-occur with severe depressive disorders [21,22].
At this point, it is appropriate to discuss the case of the wide-dynamic-range (WDR) neuron, a self-stimulating type of interneuron. As discussed, interneurons are found in the dorsal horn of the spinal cord and may act to facilitate or inhibit the transmission of nerve impulses, depending upon the receptors and neurotransmitters present. WDR neurons are associated with chronic pain states and are triggered by glutamate and glycine (excitatory neurotransmitters), which in turn activate N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors [5,12,23,24]. The NMDA receptor is a channel between the extracellular fluid and intracellular fluid embedded in the cell membrane. The dorsal horn of the spine has many glutamate- and glycine-releasing interneurons, and in an excitatory state, large numbers of glutamate and glycine neurotransmitters are released from neurons [25]. They have binding sites on NMDA receptors, which allows the movement of both Na+ and Ca2+ into cells, while a comparatively small amount of K+ exits [23,25]. The NMDA receptor cannot open, even in the presence of glutamate and glycine, without first having the magnesium ion (Mg2+) blocker removed from the center of the channel. The interneurons containing the NMDA receptors, however, have other receptors, allowing them to become excited and fire an impulse. When the initial depolarization occurs, the Mg2+ obstruction is removed from the channel, and the rich environment of glycine and glutamate allows the membrane to continually depolarize. This increases the excitability of the second-order neuron, facilitating the passage of painful stimuli to the brain. As the nervous tissue becomes increasingly excited, further releases of glutamate and glycine occur, prompting more NMDA receptor opening in a positive feedback loop. This is called a windup phenomenon [26]. In other words, the area of injury becomes so excited that hyperalgesia sets in, and, in this state, even the smallest stimulus may result in severe pain. Wind-up phenomena result in a continual stimulation of neurons within the cord, with information processed there being sent to the brain. This is referred to as long-term potentiation and is quite difficult to treat [23,24].
Knowledge of these pain pathways is necessary to achieve a sense of the many sites in the central and peripheral nervous systems where pain can be treated. As specific drug classes are described, one may return to these sections for a better these diagrams to understand how and where they act in the body.
Pain pathways stimulate many areas of the brain. The brain responds by the release of many neurotransmitters and other hormones to provide a systemic response [10]. As discussed, pain impulses activate the amygdala, which triggers a sympathetic nervous system response, sometimes referred to as the "fight-or-flight" response. The release of norepinephrine and epinephrine results in, among other things, tachycardia, hypertension, and elevated blood glucose levels. Additionally, the local response to the stimulus produces the release of local neurotransmitters, such as substance P, glutamate, CGRP, and brain-derived neurotrophic factor (BDNF) [10,11]. Of perhaps greater concern is the release of cytokines, which results in a profound inflammatory response. The inflammatory response is usually highlighted by hyperalgesia (exaggerated painful response to a painful stimulus) and allodynia (painful response from a non-pain-inducing stimulus). Take the example of a minor sunburn. If the skin is reddened and inflamed, a pat on the back becomes inordinately painful (hyperalgesia) and simply wearing a shirt may be intolerable (allodynia). In addition to these responses, untreated acute pain may lead to the expression of additional FNEs and nociceptors.
Acute pain usually has an easily recognized proximate cause and can be well-localized. The possible exception is intra-abdominal or pelvic pain, in which unmyelinated nerves are responsible for most nerve impulse propagation. However, even this hard-to-localize pain is often characterized to a general area ("My stomach hurts") rather than being poorly defined ("Everything aches").
In some cases, acute pain is associated with a medical procedure, such as routine surgery. For these patients, it is possible to visualize precisely where tissues have been manipulated and thus the location of the pain. The advantage of treating this form of acute pain is that treatment can be pre-emptive, with the administration of analgesics as part of the overall management plan [27].
Chronic pain is experienced by nearly one-third of the adult population of the United States and is associated with costs of more than $600 billion per year [11]. It can result in physical and emotional disability. It has become clear that chronic pain is far different from acute pain in its experience and treatment. While acute pain is related to a specific injury site, chronic pain is often centrally mediated and can therefore occur without the stimulation of a peripheral nerve [7,11,28]. Unfortunately, while acute pain can serve some adaptive purpose in protecting the person from harm, chronic pain is maladaptive in nature and typically has no beneficial biologic or systemic significance [11].
Historically, the initial approach to diagnosis and management of pain emphasized the identification of disease, lesion, or anatomic site of the pain, without reference to the underlying neural mechanisms or the application of this to treatment considerations [29]. Evidence now strongly supports combining the conventional etiology-based approach with a mechanism-based approach that classifies pain syndromes by the type of maladaptive nervous system alteration that has developed in reaction to the original insult. This approach provides a comprehensive dual therapeutic focus that targets the pathologic sustaining mechanism of the pain as well as the original disease, lesion, or tissue injury that has been the traditional focus of pain management [29,30]. Such an approach is believed to optimize pain diagnosis and treatment by avoiding the limitations associated with the traditional etiology-based approach [31,32,33,34,35,36].
Most pain syndromes involve multiple, often overlapping, neurobiologic mechanisms determined by the stage of the disease process. Current concepts of pain classify these into three main categories: nociceptive, neuropathic, and nociplastic [37,38,39].
Nociceptive pain is a physiologic response to tissue injury, the perception that arises from intense stimulation of specialized peripheral sensory neurons (nociceptors) that respond only to noxious (pain) stimuli. Nociceptive pain is subgrouped by location of involved tissues into somatic pain (muscle or connective tissue) and visceral pain (visceral structures). Nociceptive pain is considered adaptive during tissue healing but maladaptive and pathologic when it persists after healing has occurred. For example, inflammatory pain occurs in response to tissue injury or infection that activates peripheral nociceptors and initiates the immune response. While the resultant production and recruitment of pro-inflammatory mediators to the injury site may serve to perpetuate discomfort, it also facilitates tissue repair; thus, this is considered an adaptive pain mechanism [37,38,39].
Neuropathic pain originates from peripheral or central nervous system injury. Unlike nociceptive pain, the mechanism of neuropathic pain has no adaptive function and is strictly pathologic. Acute pain from somatosensory damage is termed "acute neural injury." The term "neuropathic pain" implies pain that persists beyond the period of expected or actual tissue healing, and the underlying mechanism involves a maladaptive alteration in somatosensory nervous system function [37,38,39].
Nociplastic pain results from alterations of nociceptive processing, such as heightened central excitability and/or diminished central inhibition (i.e., central sensitization). Nociplastic pain is typically chronic and more widespread than nociceptive pain. The mechanism is poorly understood and is regarded as strictly pathologic as it lacks any evident adaptive function. Nociplastic pain can occur independently of peripheral nociceptor activity; however, some conditions involve both nociceptive and nociplastic pain mechanisms (e.g., peripheral and central sensitization) to varying degrees along a continuum. Centralized pain syndromes include conditions such as fibromyalgia, low back pain, and carpal tunnel syndrome [37,38,39,40].
The persistence of acute nociceptive, inflammatory, or neural injury pain beyond tissue healing or repair reflects ongoing nociceptive activity that has become dissociated from peripheral nociceptive input to become maladaptive. Regardless of whether acute pain originates from tissue injury, tissue infection, or peripheral nerve injury, a similar process occurs by which nociceptive, inflammatory, and neuropathic pain signals are relayed from tissue injury site to the brain. This highly intense or prolonged pain signaling can lead to profound alteration in neuronal pathways that are further "upstream" from the peripheral tissue pain origin. Among these are increased ascending pathway signaling to the brain, reduced descending inhibitory signaling, expansion of pain receptive field, and induction of spontaneous and widespread pain. The resulting peripheral and central pathway hypersensitivity represents a state of abnormal nervous system function, amplified central nervous system sensory signaling, and abnormally low threshold pain response. The pain is no longer a symptom of peripheral insult, but a disease state of the nervous system [35]. This transition from acute to chronic pain occurs in discrete pathophysiologic steps involving multiple signaling pathways [41].
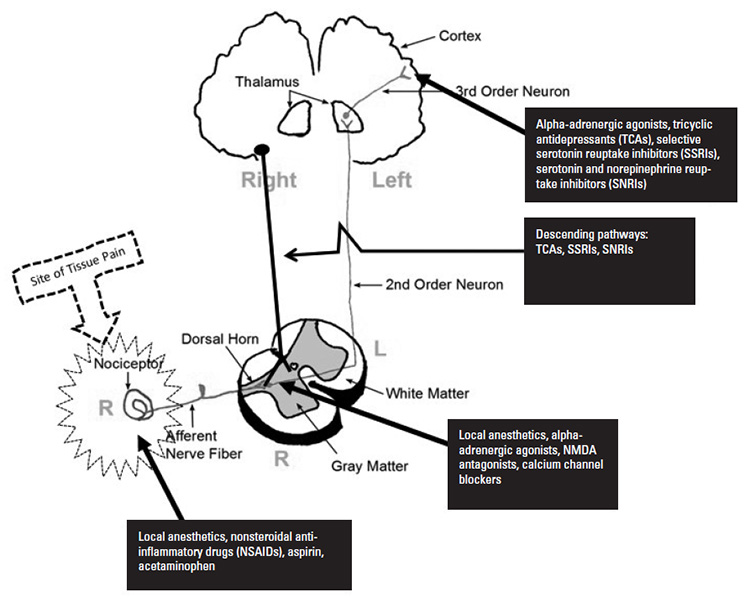
Figure 6 illustrates some of the sites and agents useful in the management of pain. Note that some agents act at more than one site along the pain pathway, and some of the agents enhance the utility of the endogenous descending pain pathways.
According to the Centers for Disease Control and Prevention, for patients already receiving opioid therapy, clinicians should carefully weigh benefits and risks and exercise care when changing opioid dosage. If benefits outweigh risks of continued opioid therapy, clinicians should work closely with patients to optimize nonopioid therapies while continuing opioid therapy. If benefits do not outweigh risks of continued opioid therapy, clinicians should optimize other therapies and work closely with patients to gradually taper to lower dosages or, if warranted based on the individual circumstances of the patient, appropriately taper and discontinue opioids.
(https://www.cdc.gov/mmwr/volumes/71/rr/rr7103a1.htm Last Accessed: October 23, 2025)Strength of Recommendation/Level of Evidence: B4 (Clinicians should help patients arrive at a decision consistent with patient values and preferences and specific clinical situations, based on clinical experience and observations, observational studies with important limitations, or randomized clinical trials with several major limitations)
Opioid analgesics produce therapeutic and side effects by mimicking endogenous opioid activity, although some opioids produce analgesia by activity outside the opioid receptor complex. Opioids widely differ in levels of affinity and activation of opioid receptor subtypes. In addition, inter-individual variation in analgesic response and side effects is significant, largely driven by genetic factors [42]. The complex interaction between unique opioid properties and individual patient characteristics dictates that a patient-tailored approach is required for opioid selection, dose initiation, and titration to optimize safety, analgesia, and tolerability.
Naturally occurring opioid compounds are produced in plants (e.g., opium, morphine) and in the body (the endogenous opioids). Endogenous opioids are peptides that bind opioid receptors, function as neurotransmitters, and help regulate analgesia, hormone secretion, thermoregulation, and cardiovascular function. The three primary endogenous opioid peptide families are the endorphins, enkephalins, and dynorphins, and the three primary opioid receptor types are mu, kappa, and delta [44,45]. A quick overview of this complex pain modulation system is helpful in understanding how opioid analgesics work.
Endogenous opioid peptides are neurotransmitter molecules in the opioid receptor complex that produce specific physiologic effects determined by neuronal distributions of the activated opioid receptor type. The endogenous opioid peptides are cleaved from the pro-hormone precursors proenkephalin, pro-opiomelanocortin, and prodynorphin. The endogenous delta opioid receptor peptides are met-enkephalin and leu-enkephalin, cleaved from proenkephalin. Prodynorphin gives rise to kappa opioid receptor agonists dynorphin A and B. Pro-opiomelanocortin encodes the peptide beta-endorphin, which has agonist activity at all three classical opioid receptors. Some endogenous opioid ligands lack specificity for opioid receptor subtypes, such as b-endorphin and the enkephalins [5,46].
Endorphins
Endorphins are synthesized in the hypothalamus and the pituitary gland. Pain, strenuous exercise, excitement, and orgasm stimulate their release, binding, and activation. Endorphins are popularized as the "natural pain killers" from their ability to induce analgesia and a general feeling of well-being. They are thought to largely mediate analgesia from acupuncture, massage, hydrotherapy, and transcutaneous electrical nerve stimulation therapy [49].
Dynorphins
Dynorphin peptides are synthesized from the precursor pro-dynorphin and have primary affinity and binding at the kappa opioid receptor. Dynorphins are distributed throughout the CNS, with highest concentrations in the brain stem, hypothalamus, and spinal cord. Their physiologic actions are diverse, and their primary function is the modulation of pain response, appetite and weight, circadian rhythm, and body temperature. Dynorphins are linked to stress-induced depression and drug-seeking behavior, and drugs that inhibit dynorphin release are under evaluation for possible use in the treatment of depression related to drug addiction [49].
Enkephalins
Enkephalin peptides, derived from pro-enkephalin, are located throughout the brain and spinal cord and are involved in regulating nociception. Enkephalins inhibit neurotransmission in pain perception pathways, reducing the emotional and physical impact of pain. Enkephalins also reside in the gastrointestinal (GI) tract, where they help regulate pancreatic enzyme secretion and carbohydrate metabolism [49].
Opioid receptors are expressed throughout the CNS and PNS on key nodes within the pain pathway and are highly concentrated in areas involved with integrating pain information [50]. Opioids vary greatly by receptor affinity, binding, and activity and can bind to produce agonist, partial agonist, or antagonist receptor activity [44]. As noted, the analgesic activity and the side effects result from mimicry of endogenous opioids, achieved by the beta-phenylethylamine group moiety shared by endogenous and exogenous opioid receptor ligands that facilitate opioid receptor binding [51].
Mu Opioid Receptors
Mu receptors are the primary mediators of analgesia produced by opioid analgesics in clinical use. Their greatest CNS concentration is in the thalamus, medulla, periaqueductal gray area, neocortex, amygdala, dorsal horn, inferior and superior colliculi, and brain stem [44,49,52]. PNS occupancy includes the peripheral sensory neuron dorsal root ganglion, stomach, duodenum, jejunum, ileum, and proximal and distal colon. Mu receptors in non-neural tissue are found in the vascular and cardiac epithelium, keratinocytes, vas deferens, and Sertoli cells [53].
Mu opioid receptors in the amygdala and nucleus accumbens mediate opioid reward response (e.g., euphoria). In this brain region, opioids bind to and activate mu receptors, which inhibit gamma-aminobutyric acid (GABA) to increase dopamine transmission [50]. Mu opioid receptors broadly distributed in the limbic system mediate emotional response to pain and analgesia. In the medial thalamic nuclei, they relay spinothalamic inputs from the spinal cord to the cingulate gyrus and limbic structures [54].
Kappa Opioid Receptors
Kappa opioid receptors bind dynorphin as the primary endogenous ligand. In the CNS, they are highly concentrated in the caudate-putamen, nucleus accumbens, amygdala, brain stem, neural lobe of the pituitary gland, and hypothalamus. In the PNS, these receptors are found in the sensory neuron dorsal root ganglion, stomach, duodenum, jejunum, ileum, and proximal and distal colon. They are primarily found in the limbic system, brain stem, and spinal cord. Their major effects include spinal analgesia, sedation, dyspnea and respiratory depression, dependence, and dysphoria [53]. The kappa opioid receptor subtype k3 is considered the primary analgesic mediator [55].
Delta Opioid Receptors
Delta receptors are mostly confined to CNS structures of the pontine nuclei, amygdala, olfactory bulbs, and deep cortex, but are also found in the GI tract and the lungs. They mediate spinal and supraspinal analgesia and the psychomimetic and dysphoric effects of opioid analgesics [49,56].
Other Potential Opioid Receptors
Other opioid-like receptors have been identified in the CNS, including the opioid receptor like-1 (ORL-1). In contrast to the classic opioid receptors, the ORL-1 receptor is insensitive to the opioid antagonist naloxone. Opioids can bind to and activate the toll-like receptor 4 (TLR4), an innate immune pattern-recognition receptor [50].
Opioid analgesia results from a complex series of neuronal interactions, largely mediated by the high density of opioid receptors in the dorsal horn of the spinal cord and in subcortical regions of the brain [46]. The analgesic effects of opioids result from two general processes: 1) direct inhibition of ascending transmission of pain signaling from the dorsal horn of the spinal cord, and 2) activation of descending pain control circuits from the midbrain to the dorsal horn of the spinal cord [49]. All three opioid receptor types mediate spinal analgesia. Supraspinal analgesia is primarily mediated by mu opioid receptor subtype 1. Opioid receptors are coupled to the superfamily of inhibitory G proteins. Receptor activation inhibits adenylate cyclase, reducing generation of cyclic adenosine 3,5 monophosphate and other second messengers. Potassium conduction is activated, inhibiting calcium influx to hyperpolarized target cells and reducing their response to depolarizing pulses. Neurotransmitter release is inhibited, and generation of postsynaptic impulses is decreased [46,50].
Although drugs such as morphine are highly selective for mu opioid receptor and bind multiple mu receptor subtypes, mu opioid agonists greatly differ by interaction with different receptor variants and other opioid and non-opioid receptors [45]. A pharmacologically and clinically relevant classification approach is classifying opioid agents by functional interaction as mu receptor agonists, partial agonists, mixed agonists-antagonists, or antagonists (Table 3) [57,58].
COMMONLY USED OPIOIDS
| Drug | Functional Category | Route(s) | Comparison to Morphinea | ||
|---|---|---|---|---|---|
| Morphine | Mu receptor agonist | IM, IV, PO, inhaled vapors | 1 | ||
| Hydromorphone (Dilaudid) | Mu receptor agonist | PO, SQ, IM, IV | 10 | ||
| Fentanyl (Actiq, Sublimaze) | Mu receptor agonist | PO, IV, buccal film, transdermal patch | 100 | ||
| Oxycodone (Roxicodone, OxyContin) | Mu receptor agonist | PO | 1.5 | ||
| Tramadol (Ultram, ConZip) | Mu receptor agonist | PO, IV | 0.1 | ||
| Hydrocodone (Hysingla ER) | Mu receptor agonist | PO | 1 | ||
| Oxymorphone (Numorphan) | Mu receptor agonist | PO, SQ, IM, IV | 0.3 | ||
| Meperidine (Demerol) | Mu receptor agonist | PO, IM, IV | 0.1 | ||
| Methadone (Methadose) | Mu receptor agonist | PO, SQ, IM, IV | Dose dependent | ||
| Codeine (Codeine) | Mu receptor agonist | PO | 0.17 | ||
| Buprenorphine (Belbuca, Butrans, Sublocade) | Partial mu receptor agonist | PO, buccal film, transdermal patch, IM, IV | 30 | ||
| Butorphanol (Stadol) | Mixed agonist/antagonist | Nasal spray, IM, IV | 2 | ||
| Nalbuphine (Nubain) | Mixed agonist/antagonist | SQ, IM, IV | 1 | ||
| Sufentanil (Dsuvia) | Mu receptor agonist | IV | 1,000 | ||
| Naloxone (Narcan) | Antagonist | IV, IM, SQ, nasal spray | -- | ||
| |||||
Spinal Level
The spinal cord dorsal horn is a primary analgesic site of opioids and is densely populated with mu (70%), delta (20%), and kappa (10%) opioid receptors. Opioid receptors are localized on presynaptic afferent fibers, interneurons, and postsynaptic projection neurons [50]. Opioids bind to and activate mu receptors, which inhibit the release of pain mediators such as substance P, glutamate, and nitric oxide from nociceptive afferent neurons. Spinal level analgesia appears to elevate pain thresholds [46].
Supraspinal Level
At supraspinal levels, opioids produce analgesia by attenuation of the subjective evaluation of pain. After morphine is given for severe pain, patients report pain but without the associated anguish and distress. Conscious awareness and pain response are retained but modified by changes in emotional response to pain, mediated in part through opioid receptors in the limbic system [46].
Opioid receptors are highly concentrated in the medial thalamus, where incoming sensory information associated with intense and deep pain is filtered and then relayed to the cerebral cortex. This opioid effect on medial thalamus pain signal filtering greatly contributes to analgesia [46].
Opioid receptors are highly localized in subcortical brain regions where descending pain-modulating pathways originate. Normally, these pathways are inhibited by GABAergic neurons that project to descending inhibitory neurons of the brain stem. Opioid analgesics bind to and activate mu receptors on GABAergic neurons; this inhibits GABA to activate descending pain-modulating pathways [46,50]. In addition, opioids activate ascending serotonin/norepinephrine pathways that project to forebrain centers to regulate the emotional response to pain [44].
The greatest factor that contributes to opioid analgesia is concentration of the drug on the mu receptor, which can be altered by pharmacokinetic processes that influence plasma concentration of the opioid by impacting its absorption, distribution, metabolism, or excretion. Intrinsic properties of the opioid, such as lipid solubility, also contribute to opioid receptor concentration [59].
Neuropathic Pain
Opioid analgesics have historically been considered less effective in neuropathic pain, but more recent evidence provides some support for their use. The extent of neuropathic pain reduction correlates with the duration of opioid therapy, possibly accounting for the mixed results in short-term studies [60,61]. A 2011 study discovered previously unknown mu and kappa receptor expression on numerous peripheral tissues, immune cells, and joint capsules/synovium. The administration of opioids by injection into painful peripheral tissue sites results in pain relief in the absence of CNS activity, which supports the existence of localized peripheral opioid receptors [62].
Opioid effectiveness in neuropathic pain may be influenced by the capacity to inhibit voltage-gated sodium channels and individual channel type. Buprenorphine is more effective in blocking sodium channels than meperidine, lidocaine, and bupivacaine, possibly from greater lipophilicity, as this is a major factor in local anesthetic potency [61]. Sufentanil, fentanyl, and tramadol, but not morphine, are effective in blocking the voltage-gated sodium channel 1.2 (Nav 1.2) and may have greater clinical effect in some forms of neuropathic pain [63].
Inflammation enhances opioid anti-nociceptive action by peripheral mechanisms that activate during later (but not early-stage) inflammation, suggesting that timing of opioid administration contributes to analgesic efficacy in inflammatory pain [62]. Opioids are also effective in reducing the "air hunger" of dyspnea in patients suffering from cancer or respiratory or cardiovascular insufficiency [44].
A fourth group of opioids, opioid antagonists, bind and inactivate opioid receptors. Naltrexone and naloxone have traditionally been used to reverse potentially fatal overdose from opioid receptor agonists such as morphine or heroin. Opioid agonist molecules on mu opioid receptor are displaced, agonist effects on mu opioid receptor are abruptly halted, and opioid-dependent patients rapidly experience full alertness, analgesic loss, and opioid withdrawal [64].
Clinical trials with low-dose naltrexone have found unexpected and paradoxical enhancement rather than blockade of analgesia when co-administered with morphine and other opioid agonists in postoperative pain or severe intractable pain. Other evidence suggests analgesic efficacy as monotherapy in Crohn disease, irritable bowel syndrome, and fibromyalgia [65]. These findings led to the development and introduction of the peripheral-acting mu receptor antagonists alvimopan, methylnaltrexone, naloxegol, and naldemedine for severe opioid-induced constipation [58,66,67].
In addition to opioid-induced constipation, opioid antagonists are U.S. Food and Drug Administration (FDA)-approved for the treatment of alcohol and opioid use disorder (naltrexone 50–100 mg/day oral) and opioid overdose (naloxone 0.4–1.0 mg/dose IV or IM). In pain medicine, the dose ranges of naltrexone and naloxone are substantially lower. Of the two, naltrexone is much more widely used, and published pain medicine studies have used dose ranges of 1–5 mg (termed "low-dose") or <1 mg in microgram amounts (termed "ultra-low-dose") [65]. For example, case studies have reported dramatic improvement in refractory pain with intrathecal administration of an opioid agonist combined with ultra-low-dose naloxone in the low nanogram range [68].
The mechanism of low-dose and ultra-low-dose opioid antagonists is not fully known and is the subject of investigation [65]. One explanation describes a sequential action, whereby binding and inhibition first occurs at excitatory receptors, followed by binding at inhibitory receptors. This decrease in excitation facilitates a broader clinical expression of inhibitory function, which potentiates analgesia and reduces adverse effects. For example, with opioid-induced hyperalgesia, ultra-low-dose naltrexone appears to act through excitatory blockade to promote analgesia and tolerability [58,69,70].
NSAIDs alleviate pain by inhibiting the conversion of arachidonic acid to prostaglandins catalyzed by COX isozymes. Nonselective NSAIDs inhibit COX-1 and COX-2 and include ibuprofen, aspirin, and naproxen. The nonselective action inhibits the formation of both gastroprotective-mediating prostaglandins and pain-promoting prostaglandins, increasing the risk of serious toxicities such as GI ulceration and bleeding. This prompted the development of selective COX-2 inhibitors, which produce fewer GI side effects but are linked with an increased risk of cardio-renal morbidities [71]. To mitigate risk of GI adverse events, proton pump inhibitors are recommended for use in some patients using NSAIDs [72].
Acetaminophen is available over the counter and is also included in combination with many prescription opioids. Analgesia is achieved through central but not peripheral inhibition of prostaglandin. Although effective in mild pain, acetaminophen is not anti-inflammatory. The side-effect profile is relatively benign with intermittent use at recommended labeled dosing, but long-term or high-dose use can be hepatotoxic, and the daily dose should never exceed 4 g. Acetaminophen is recommended over NSAIDs as an analgesic in patients with GI, renal, or cardiovascular comorbidity [73].
While beneficial in the management of pain, some patients with reactive airway disease may develop bronchospasm, although this adverse effect is rare [5]. In addition, prostaglandins are vasodilators. A constitutive level of circulating prostaglandin is necessary to maintain adequate vasodilation in the afferent limb of the glomerulus to assure renal blood flow in the kidney. Overuse of NSAIDs may result in decreased glomerular blood flow, resulting in decreased elimination of toxins from the body and, in especially severe states, renal failure [5]. The most commonly used NSAIDs and acetaminophen with corresponding brand names and routes of administration are listed in Table 4[58,75].
COMMONLY USED NONSTEROIDAL ANTI-INFLAMMATORY DRUGS (NSAIDs) AND ACETAMINOPHEN
| Drug | Route(s) |
|---|---|
| Meloxicam (Anjeso) | IV, PO |
| Ketorolac (Toradol) | PO, IM, IV, eye drops, nasal spray |
| Ibuprofen (Motrin, Advil) | PO, IV |
| Diclofenac (Cataflam, Voltaren) | PO, IM, IV, eye drops, topical gel |
| Naproxen (Aleve, Anaprox) | PO |
| Celecoxib (Celebrex, Elyxyb) | PO |
| Aspirin | PO |
| Acetaminophen (Tylenol) | PO, IV, rectal |
Local anesthetics prevent the generation and propagation of nerve impulses in response to painful stimuli. The basic chemical structure of a local anesthetic consists of an aromatic ring (which enhances lipid solubility) and an intermediate ester or an amide chain and a terminal amine [8]. As such, all of these agents are classified as either an ester type or an amide type. The type of anesthetic used and the inclusion of a vasoconstrictor (e.g., epinephrine) will influence the duration of action. Certain factors, such as the presence of active infection in the area to be anesthetized, heightened patient anxiety, or inaccurate deposition of the agent, may affect the ability of a local anesthetic to achieve the appropriate level of anesthesia.
The local anesthetics lidocaine and bupivacaine block Na+ influx of voltage-gated ion channels in afferent neuron terminals, inhibiting depolarization and generation of action potentials, resulting in the transmission of fewer nociceptive impulses to the spinal cord. In clinical application, topical lidocaine is used for neuropathic pain to block hyperactive sodium ions in damaged peripheral nerves and inhibit transmission of ectopic impulses to the dorsal horn. This action interferes with peripheral and central sensitization and maladaptive neuroplasticity [71,76].
Capsaicin defunctionalizes nerve fiber terminals through multiple mechanisms to produce analgesia. The initial reduction in neuronal excitability and responsiveness result from inactivation of voltage-gated sodium channels and direct desensitization of plasma membrane TRPV1 receptors. This is followed by extracellular Ca2+ entry of TRPV1 and release from intracellular stores to overwhelm the TRPV1 receptor intracellular Ca2+ buffering capacity, subsequent activation of calcium-dependent proteases, and cytoskeleton breakdown [77,78]. The persistent effect involves extracellular Ca2+ entry of TRPV1 and release from intracellular stores to overwhelm TRPV1 receptor intracellular Ca2+ buffering capacity, subsequent activation of calcium-dependent proteases, and cytoskeleton breakdown [77,78]. Capsaicin is available as a high-potency (8%) patch and as a lower-concentration cream. A single 60-minute application may provide up to 12 weeks of analgesia [76]. Capsaicin may initially cause pain because substance P is released from nociceptive terminals to initiate nociceptive firing. The analgesic response follows as nociceptive terminals desensitize to elevate pain threshold [79].
The use of local anesthesia has become more popular and may be more precisely administered being guided by ultrasonography. Ultrasound technology allows for optimized needle placement, resulting in fewer failed blocks and lower doses. These blocks can be performed before having a procedure performed. When administered before surgery, local anesthetic blocks allow for lower doses of anesthetic agent, along with prolonged postoperative pain relief. In one study, when a local anesthetic block was provided in addition to typical analgesic therapy following total knee replacement, morphine doses, pain scores, and nausea were all significantly decreased compared with those who received usual treatment [80].
However, local anesthetics are not without drawbacks, and overdose resulting in local anesthetic systemic toxicity (LAST) is a concern [81]. Because local anesthetics are designed to cross phospholipid membranes, they easily enter the brain and heart, where the blockade of ion channels can have adverse effects. Normally, the first symptoms in patients who are conscious are a metallic taste and/or ringing in their ears (tinnitus). If left untreated, this can progress to excitatory symptoms, followed by drowsiness, coma, and even death [81,82]. Table 5 provides key information about commonly used local anesthetics, including maximum doses.
MAXIMUM DOSES OF LOCAL ANESTHETICa
| Local Anesthetic | Ester or Amide | Maximum Dose Per Kilogram Plain | Maximum Dose Plainb | Maximum Dose Per Kilogram with Epinephrine | Maximum Dose with Epinephrineb | ||
|---|---|---|---|---|---|---|---|
| Bupivacaine (Marcaine) | Amide | 2 mg/kg | 175 mg | 3 mg/kg | 225 mg | ||
| Levobupivacaine (Chirocaine) | Amide | 2 mg/kg | 200 mg | 3 mg/kg | 225 mg | ||
| Lidocaine (Xylocaine) | Amide | 5 mg/kg | 350 mg | 7 mg/kg | 500 mg | ||
| Mepivacaine (Carbocaine) | Amide | 5 mg/kg | 350 mg | 7 mg/kg | 500 mg | ||
| Ropivacaine (Naropin) | Amide | 3 mg/kg | 200 mg | 3 mg/kg | 500 mg | ||
| Prilocaine (Citanest) | Amide | 6 mg/kg | 400 mg | 8 mg/kg | 250 mg | ||
| Procaine (Novocaine) | Ester | 7 mg/kg | 1,000 mg | 10 mg/kg | 600 mg | ||
| Tetracaine (Amethocaine) | Ester | 0.2 mg/kg | 20 mg | N/A | 1,000 mg | ||
| |||||||
The closer to the vasculature the site of injection is, the more likely that the local anesthetic will undergo rapid vascular uptake and result in adverse effects. Mixing the agent with epinephrine, a vasoconstrictor, decreases uptake, thus prolonging the anesthetic effect and decreasing the likelihood of complications. As this is the case, it is possible to give more local anesthetic when it is mixed with epinephrine [82,83,84].
In the event of an accidental overdose, there is a specific protocol for treatment [81,82]. As soon as LAST is detected, the patient should be administered an IV bolus injection of 20% lipid emulsion 1.5 mL/kg-1 over one minute. An infusion of 20% lipid emulsion should be started at a dose of 15 mL/kg-1/hour. If after five minutes cardiovascular stability is not restored or the patient further deteriorates, a maximum of two repeat boluses may be administered, with five minutes between injections. At the same time, the infusion rate may be doubled. The infusion should continue until the patient has stabilized or the maximum dose of emulsion (12 mL/kg-1) has been given. The use of lipids to rescue these patients is a relatively new development, so review is important for those in clinical areas with high use of local anesthetics [85].
The gabapentinoids, gabapentin and pregabalin, are widely used in the management of both postoperative and chronic pain relief. Their names may give the impression they interact with gamma-amino butyric acid (GABA), but this is not the case [86,87]. Gabapentin and pregabalin are anticonvulsants that are also effective in a wide range of neuropathic pain conditions. Their mechanism of action involves selective binding to and blockade of the α2δ1 subunit of voltage-gated calcium channel in various brain regions and the superficial dorsal spine. This inhibits the release of glutamate, norepinephrine, and substance P to decrease spinal cord levels of neurotransmitters and neuropeptides [76,88,89]. The binding affinity of pregabalin for the calcium channel α2δ1 subunit is six times greater than gabapentin, which is reflected in the greater efficacy of pregabalin at lower doses. Because gabapentin possesses a shorter half-life and nonlinear absorption, pregabalin is easier to titrate and better tolerated [89].
While having a long history in the treatment of chronic pain, the use of these agents to prevent postsurgical pain is relatively new. In one study of 90 women scheduled for abdominal hysterectomy, a control group was compared to groups receiving either 300 mg pregabalin or 900 mg gabapentin administered one to two hours prior to surgery [86]. The average time until first request for analgesia was 31 minutes in the pregabalin group, 16 minutes in the gabapentin group, and 7 minutes in the control group. There was no difference in demographic variables, including length of surgery, across the three groups. In this case, preoperative administration of a gabapentinoid was shown effective in lengthening the duration of analgesia [86].
The locus coeruleus is activated during normal responses to painful stimuli. However, in patients with chronic pain, stimuli inhibit rather than activating the locus coeruleus, dampening analgesic response [90]. When gabapentin is administered to these patients, glutamate is released in the brain, which in turn stimulates the locus coeruleus, restoring its analgesic function [91].
While more commonly associated with the autonomic nervous system and its functions, alpha-adrenergic agonists can also function in the relief of pain, as well as decreasing the sympathetic side effects which accompany pain, including hypertension and tachycardia. Antinociceptive activity of the α-2 adrenoceptor agonists clonidine and tizanidine includes modulating dorsal horn neuron function and norepinephrine and 5-HT release, potentiating mu-opioid receptors, and decreasing neuron excitability through calcium channel modulation [92]. Clonidine is available as a transdermal patch for use in neuropathic pain states. Local use enhances release of endogenous enkephalin-like substances. Intrathecal or epidural administration with opioids and/or local anesthetics is favored in treating neuropathic pain because the synergistic effect improves pain control. Tizanidine is used as a muscle relaxant and antispasticity agent; its use in the management of musculoskeletal pain is off label [76,79].
Dexmedetomidine was originally approved as a short-term sedative analgesic for mechanically ventilated patients in the intensive care unit [93]. Dexmedetomidine is far more selective as an alpha-adrenergic agonist and has the same central action around the locus coeruleus [93]. As time passed since its introduction, the use of dexmedetomidine has increased, especially among patients with comorbidities (e.g., heart and vascular disease, morbid obesity). Its cardiovascular stability, along with its minimal effect on respiratory drive after the infusion is terminated, have made this agent popular in both the intensive care unit and the operating room. Aside from its use as a sedative or aesthetic agent, use of dexmedetomidine has been explored in patients with refractory end-of-life pain. In a case study, a male patient, 58 years of age, with chronic pancreatitis secondary to alcoholism reported inadequate pain relief despite receiving a combination of oxycodone, nortriptyline, and lorazepam. Increased inpatient intravenous opioids and ketamine still brought the patient no relief, and dexmedetomidine was attempted as a last resort. An infusion of dexmedetomidine brought the patient's pain under greater control, to the extent that he was able to sit in a recliner and visit with family [94]. Based on this and other reports, dexmedetomidine is being explored as a possible option in palliative care.
Alpha-adrenergic agonists are also found in the area of the brain where projections from the locus coeruleus inhibit an inhibitory portion of the brain responsible for arousal. When alpha-adrenergic agonists act in this area of the brain, they block the ability of the nerves projecting from the locus coeruleus to inhibit the second-order neuron. Thus, these agents, in sufficient doses, render the patient somewhat drowsy [95]. This is important to remember, as the drowsiness associated with the original alpha-adrenergic agonists made their use problematic.
Anesthetics are powerful agents typically used in the operating room to reduce the capacity for consciousness and diminish the pain associated with surgery. Two examples are ketamine and nitrous oxide. These agents are quite different; the first is an injectable dissociative anesthetic with variable effects depending on the dose, and the second is an inhaled vapor with profound analgesic effects [18,96,97].
Ketamine is a phencyclidine anesthetic given parenterally, neuraxially, nasally, transdermally or orally in subanesthetic doses to alleviate a variety of pain conditions, including severe acute pain, chronic or neuropathic pain, and opioid tolerance [79]. The mechanism of analgesic effect primarily involves NMDA receptor inhibition. Thus, patients with NMDA-mediated central sensitization are likely to realize significant benefit from treatment with ketamine. Ketamine also has activity on nicotinic, muscarinic, and opioid receptors and exerts both anti-nociceptive and anti-hyperalgesic effects, with the latter produced at lower dose ranges [98].
The American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists recommend that subanesthetic ketamine infusions be considered for patients undergoing painful surgery and patients undergoing surgery who are opioid-dependent or opioid-tolerant.
(https://rapm.bmj.com/content/rapm/43/5/456.full.pdf Last Accessed: October 23, 2025)Strength of Recommendation: B (There is high certainty that the net benefit is moderate, or there is moderate certainty that the net benefit is moderate to substantial.)
Ketamine is one of very few therapies demonstrating substantial and durable pain reduction of treatment-refractory chronic regional pain syndrome [99]. Potentially distressing adverse reactions (e.g., hallucinations, disturbing dreams, out-of-body experiences) and unwanted changes in mood, perception, and intellectual performance have limited its clinical use in pain control. However, trials have effectively controlled these side effects with high-dose co-administration of midazolam or lorazepam combined with either clonidine or ondansetron [100,101].
Ketamine, however, also has its down sides. One of the most concerning is the formation of psychotomimetic behaviors, and the presence of hallucinatory phenomena after its administration. Fortunately, these are dose dependent in nature and rarely occur at the doses required to treat pain [102,103]. It has also become a drug of abuse and misuse. Most notoriously, ketamine became known as a "date-rape drug," because it was administered in drinks to unknowing victims who were subsequently sexually assaulted by their predators. Because ketamine causes amnesia, victims have little or no memory of what occurred to them, although they often experienced after-effects, such as pain. As a result of this growing criminal use, Congress passed the Drug-Induced Rape Prevention and Punishment Act of 1996. During this period and the decade following, there was increased awareness of the dangers of ketamine and other drugs that were used in a similar manner, such as flunitrazepam (Rohypnol) and gamma hydroxybutyric acid (GHB) [104]. As a result, ketamine developed a stigma, and this negative view may persist in many minds.
Today, ketamine is increasingly being used to treat patients with treatment-refractory major depressive disorder, which frequently co-occurs in those with chronic pain. The agent appears to actually increase the size and volume of the hippocampus, thus treating the cause of depression [18,105]. In patients who are imminently suicidal, short-duration doses have been found to significantly reduce suicidal ideation [106].
Nitrous oxide (chemical formula N2O) is a component familiar to many, as it is commonly used today to facilitate comfort and address anxiety in dental settings. Historically, it has been used in both dental and medical interventions. Nitrous oxide is a compressed gas and is one of the oldest anesthetic agents in use, with its origins dating back to 1772 [96]. Unlike the inhaled hydrocarbons commonly used as part of a general anesthetic, nitrous oxide has potent analgesic properties. It is thought that nitrous oxide works to enhance the endogenous descending pathways to the alpha-2 and GABA-A neurons in the spinal cord, decreasing the ability of the second-order neurons to depolarize and carry painful stimuli to the brain. When administering nitrous oxide, it is crucial to ensure that oxygen is added to prevent the administration of 100% nitrous oxide to the patient, which would rapidly result in hypoxia and death. All certified nitrous oxide delivery devices have lockout systems to preclude this from happening.
Nitrous oxide is given as a percentage of total inhaled gas flow. The route of administration is inhalation via a mask secured to the patient's nose. For analgesic purposes, the concentration is typically 50% to 70% nitrous oxide with oxygen. Onset of action can occur in as quickly as 30 seconds, with the peak effects seen in five minutes or less. Nitrous oxide diffuses into the alveolus very quickly, accounting for its rapid uptake and circulation to the brain. Nitrous oxide is not metabolized in the body. It is eliminated via respiration within minutes after 100% oxygen is inhaled at the conclusion of the intervention [107].
Repeated doses can be problematic, as extended use of nitrous oxide has been linked to vitamin B12 deficiency [108]. As such, serum vitamin B12 level may need to be measured before and after treatment. Of more concern is the continuous exposure of hospital or clinic staff to chronic low doses of nitrous oxide [96]. Limits of nitrous oxide in the ambient environment are strict and tightly regulated by the by the National Institute for Occupational Safety and Health (NIOSH) [109]. The maximum recommended level of exposure is 25 parts per million per procedure over an eight-hour period [109]. Sufficient fresh air flow in the procedural area is required, along with a secure fitting of the delivery mask. Nitrous is highly diffusible and will enter into closed spaces very easily.
For a short period, prehospital paramedics were using 50% nitrous oxide as an analgesic during stabilization and transport of patients to the hospital; however, this use did not gain traction, and nitrous oxide is not a universal requirement for emergency medical vehicles [110].
As with other analgesics, nitrous oxide tanks should be secured, as there is a potential for abuse and diversion, particularly in locations in which small tanks are used and can easily be removed and transported. Nitrous oxide also enhances combustion, so care should be taken when using it around lasers and electric cautery. This agent is associated with increased rates of postoperative nausea and vomiting, but the risk decreases with the duration of administration.
Antidepressants, including tricyclic antidepressants (TCAs), selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), and monoamine oxidase inhibitors (MAOIs), are now a mainstay of pain management. Each of these agents increases the circulating level of neurotransmitters (e.g., norepinephrine, serotonin, dopamine, acetylcholine) in the brain [111]. Note that while these agents have been used for pain management in some cases for decades, this use is often still considered to be off label [58,111].
Antidepressants act in the brain at the periaqueductal grey, the amygdala, the prefrontal cortex, the thalamus, and the somatosensory cortex, among other places [112]. However, they also work in the periphery, primarily by blocking voltage-gated Ca2+ channels, especially in the dorsal horn of the spinal cord. As discussed, when Ca2+ cannot enter a neuron, then exocytosis of neurotransmitters onto the receptors of the next order neuron cannot take place. This, in turn, blocks the transmission of the action potential and thus the painful stimulus [112]. Antidepressants also increase the effectiveness of endogenous GABA, an inhibitory neurotransmitter. The various antidepressant classes have different effects on pain pathways.
TCAs are widely used in neuropathic pain. A TCA's mechanism involves blocking pre-synaptic reuptake of norepinephrine and serotonin; inhibition of neuronal membrane ion channels by reducing neuronal influx of calcium or sodium ions; and activity with adenosine and NMDA receptors [79]. A primary site of analgesic action is the descending modulatory pathway, where monoamine reuptake inhibition elevates norepinephrine and serotonin levels to enhance endogenous nociceptive inhibition. The secondary amines nortriptyline and desipramine are favored over the tertiary amines amitriptyline and imipramine due to more benign side effect profiles [113,114]. Amitriptyline is often the treatment of choice for neuropathic pain [79]. Unfortunately, TCAs have numerous side effects, including xerostomia (dry mouth), tachycardia, urinary retention, and drowsiness [111].
SSRIs were designed to treat depression by increasing the amount of circulating serotonin in the brain. This increased amount of serotonin results in down-regulation (decreased number and density) of the 5-HT receptors, which allows for an increased firing of serotonergic neurons in the brain [111]. Compared with the other antidepressants, SSRIs have limited utility in treating pain and are seldom prescribed for this purpose.
The dual serotonergic and noradrenergic re-uptake inhibitors (SNRIs) duloxetine, venlafaxine, and milnacipran are widely used in the treatment of neuropathic pain conditions. Duloxetine is used in painful diabetic neuropathy, with demonstrated efficacy at 60–120 mg/day. Venlafaxine behaves like a SSRI at doses of ≤150 mg/day and like an SNRI at doses >150 mg/day; a dose ≥150 mg/day is often necessary to achieve pain control [76]. Of the three available SNRIs, milnacipran has the greatest affinity for norepinephrine, duloxetine has the greatest potency in blocking serotonin, and venlafaxine selectively binds to the serotonin but not the norepinephrine transporter [115].
SNRIs are better tolerated than TCAs because they lack affinity for cholinergic, histaminic, and adrenergic receptors [89]. The anti-nociceptive effect of the SNRIs duloxetine and milnacipran primarily involves increasing serotonin and norepinephrine concentrations in descending inhibitory pain pathways, which enhances the suppression of afferent spinal inputs and reduce pain [113].
MAOIs work by irreversibly degrading the monoamine oxidase enzymes responsible for degrading norepinephrine. These agents, however, have numerous side effects, including hypotension, dizziness, headache, xerostomia, palpitations, and weight gain [116]. One potential issue is the interaction of MAOIs with tyramine and tryptophan. With oral ingestion, MAOIs inhibit the catabolism of dietary amines. When foods containing tyramine (e.g., red wine, aged cheeses and meats, soy sauce, tap beer, smoked or pickled fish, sauerkraut) are consumed, the individual may suffer from hypertensive crisis. If foods containing tryptophan (e.g., milk, poultry, tofu, nuts, seeds) are consumed, hyperserotonemia may result. The amount required to cause a reaction varies greatly from individual to individual and depends on the degree of inhibition, which in turn depends on dosage and selectivity. These side effects limit the utility of MAOIs in pain management [116,117].
The Orthopaedic Trauma Association Musculoskeletal Pain Task Force recommends the use of multimodal analgesia (MMA) as opposed to opioid monotherapy for pain control. MMA may include NSAIDs, acetaminophen, gabapentinoids, and immediate-release opioids.
(https://journals.lww.com/jorthotrauma/fulltext/2019/05000/clinical_practice_guidelines_for_pain_management.11.aspx Last Accessed: October 23, 2025)Strength of Recommendation/Level of Evidence: Strong recommendation, moderate-quality evidence
With a clear understanding of the pharmacologic tools available to help manage pain, clinicians can begin the process of creating and supporting a pain management plan for each patient's unique needs. The World Health Organization (WHO) analgesic ladder, introduced in 1986 and disseminated worldwide, remains recognized as a useful educational tool but not as a strict protocol for the treatment of pain. It is intended to be used only as a general guide to pain management [118]. The three-step analgesic ladder originally intended for management of cancer-related pain designates the type of analgesic agent based on the severity of pain (Figure 7) [118]. Step 1 of the WHO ladder involves the use of nonopioid analgesics, with or without an adjuvant (co-analgesic) agent, for mild pain (pain that is rated 1 to 3 on a 10-point scale). Step 2 treatment, recommended for moderate pain (score of 4 to 6), calls for a weak opioid, which may be used in combination with a step 1 nonopioid analgesic for unrelieved pain. Step 3 treatment is reserved for severe pain (score of 7 to 10) or pain that persists after Step 2 treatment. Strong opioids are the optimum choice of drug at Step 3. At any step, nonopioids and/or adjuvant drugs may be helpful.
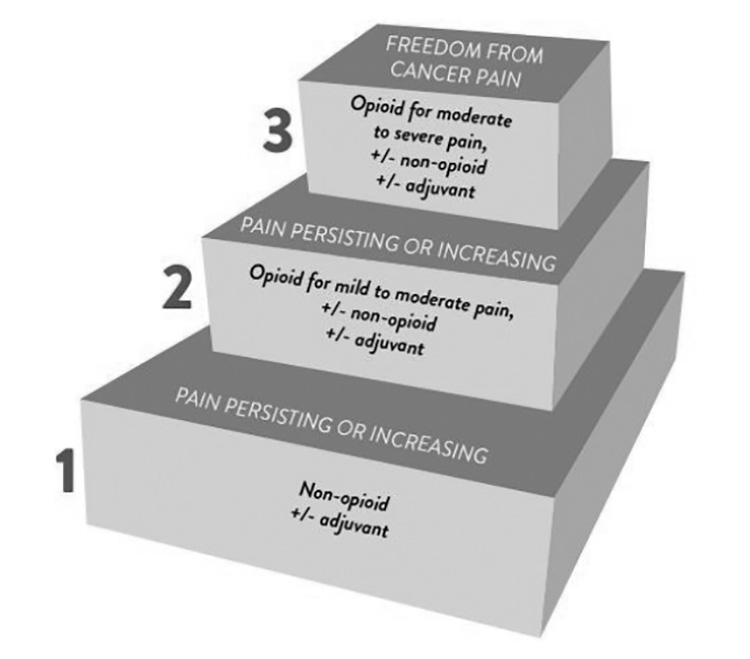
The pharmacologic treatment of pain involves selecting the right drug(s) at the right dose, frequency, and route, and managing side effects. As with any healthcare action, it is vital to assess patients and to attempt to identify underlying cause(s) prior to the initiation of treatment. Specific evaluative steps should be taken to determine the nature of a patient's pain and to assess the possibility and impact of adverse effects. The WHO ladder is also accompanied by guiding principles [118,119]:
Believe the patient's report of pain. This sounds simple, but it can be difficult for clinicians to avoid becoming jaded over time, especially if they care for patients in drug-seeking environments.
Initiate discussions of pain by asking specific questions and observing behaviors, such as groaning, a furrowed brow, and elevations in pulse or blood pressure.
Get the facts about the pain. A helpful mnemonic taught to prehospital providers is OPQRST:
Onset
Provocation or palliation
Quality
Region (of the body) and radiation
Severity
Timing
Evaluate the patient's psychological state.
Perform a detailed physical assessment.
Obtain further testing if one is not sure, including radiologic and laboratory tests.
With these data, the provider is now ready to plan and carry out multimodal analgesia. The following examples are presented as examples of the applicability and efficacy of multimodal approaches in research studies.
In one study, 150 patients were assessed for breakthrough pain following shoulder surgery [120]. The first group (75 patients) was given a standard course of opioid and acetaminophen combination (hydrocodone 10 mg/acetaminophen 325 mg) or 5–10 mg of oxycodone every 4 hours [120]. They also received a single-shot interscalene regional nerve block with 0.5% ropivacaine (local anesthetic). Finally, intravenous hydromorphone was also available as a rescue intervention. In the second multimodal group (75 patients), patients received preoperative 300 mg celecoxib, 600 mg gabapentin, and 1,000 mg acetaminophen. They also received the same regional nerve block. For postoperative pain, group 2 received naproxen 500 mg every 12 hours with food, gabapentin 300 mg every 8 hours, acetaminophen 1,000 mg orally or IV, followed by 500 mg orally every 6 hours. For breakthrough pain, this group could receive 5–10 mg of hydrocodone, as needed [120]. There were no differences in the demographic makeup of the two groups.
On postoperative day zero (the day of surgery), pain scores were significantly lower in group 2 when compared with the standard group [120]. On postoperative days 1 and 2, the multimodal group continued to have lower scores, but the differences were not statistically significant. Opioid consumption, measured in mg of morphine equivalent, was significantly decreased in the multimodal group on all three measurement days. The length of in-patient stay for the multimodal group was significantly lower (1.4 days +/- 0.7) compared with the opioid group (1.9 days +/- 1.1 days), resulting in an average cost savings of $1,000 for the multimodal group [120].
In this study, patients with unresectable hepatocellular carcinoma received transarterial chemoembolization, a primary palliative treatment [121]. This group of patients have chronic pain, and transarterial puncture is associated with a lower degree of surgical pain. In this example, patients are provided with a therapeutic procedure that can partially mitigate the pain and then supported with other analgesic agents. The study involved a total of 84 patients, with half assigned to the multimodal group and the other half assigned as a control group [121].
The multimodal group received 40 mg intravenous parecoxib sodium 30 minutes before the beginning of the procedure. In the control group, patients received 5 mg dezocine (an opioid) preintervention. All patients underwent a percutaneous puncture of the femoral artery after having the site numbed with 10 mL 2% lidocaine (total dose: 200 mg lidocaine). After the procedure, the multimodal group was provided with a patient-controlled analgesia pump; the intravenous pain solution was a combination of sufentanil 100 mcg and dexmedetomidine 200 mcg diluted in 100 mL normal saline. The pump was programmed to provide 2 mL of the solution (2 mcg of sufentanil and 4 mcg of dexmedetomidine) as a first dose, a background infusion rate of 2 mL/hour, and 2 mL bolus doses on demand with a lockout period of 5 minutes (maximum: about 12 doses per hour) [121]. This sufentanil dose is below the usual administered for general anesthesia. The dose of dexmedetomidine tracks closely to that needed for intensive care unit (ICU) sedation [122]. Using the two agents together provides central sedation via two routes (one from the opioid, the other from the alpha-2 agonist) while simultaneously providing pain relief throughout the spinal cord.
The control group received 100 mg flurbiprofen (an NSAID) every 12 hours for the first 48 hours postprocedure and 100 mg tramadol (a combination opioid agonist and SSRI) for breakthrough pain [121]. Mean visual analog scale (VAS) pain scores were measured at 0, 2, 4, 6, 12, 24, and 48 hours after the procedure. Patients in the multimodal group had statistically lower VAS pain scores at 0, 2, 4, 6, and 12 hours after the procedure. From a qualitative viewpoint, more than 95% of the multimodal patients reported good satisfaction with their pain control. In the control group, 69% reported good satisfaction, and 11.9% reported a "fairly bad experience" of pain control [121].
The following case examples detail how specific patients were cared for and the logic behind analgesic decision-making. This should serve as a starting point that will result in further self-exploration.
Note that this first example is directed toward the management of acute pain, and the interventions take place in the hospital or surgical facility. Many people, however, suffer chronic pain and self-medicate to treat it. In either case, multimodal analgesic techniques may still be used.
Patient A is scheduled to receive a total knee replacement arthroplasty [123]. Preoperatively, the patient is counseled regarding what pain might be expected with this surgery and how it might be treated. After the patient has been worked up, she receives acetaminophen 1,000 mg and celecoxib 400 mg by mouth [123]. This is referred to as pre-emptive analgesia and is done to ensure that the processes needed to, in this case, block the inflammatory effects of prostaglandins released by surgery are beginning to function prior to initiation of surgery [43].
The patient is next taken to a block room and receives local anesthesia in the knee area. In other cases, surgeons may inject local anesthesia at the end of the case.
During surgery, the patient receives a spinal anesthetic with local anesthesia. The anesthetic is placed in the subarachnoid space with local anesthesia. Because the local anesthetic is deposited so close to the nerves, a very small dose can provide several hours of anesthesia.
After surgery, the patient begins to receive several pain management interventions almost immediately, the first of which is cryotherapy. Next, as the body is responding with an inflammatory process releasing prostaglandins and other neurotransmitters in response to an injury (albeit a therapeutic one), the patient receives 1,000 mg of acetaminophen every six hours around the clock [123]. This patient tolerated oral analgesics, but acetaminophen can be administered intravenously for those experiencing problems with postoperative nausea and vomiting. The patient is also started on celecoxib 200 mg twice per day for up to five days. At this point, Patient A begins to question the need for NSAIDs when she is "having no pain." The nurse describes the importance of reducing inflammation in simple terms. He also explains that the long-acting local anesthetics will wear off over time, and it is important to pre-emptively control pain. Despite these interventions, some patients will experience postoperative pain exceeding the ability of NSAIDs to mitigate. For these individuals, an opioid rescue (oxycodone tablet 5 mg and intravenous hydromorphone 0.2 mg) every four hours as needed will help bring most pain under control [123].
Patient B is an elderly man (85 years of age) with chronic and unremitting pain. Initial assessment of the patient's pain remains important. While Patient B is experiencing chronic pain, he may also have an unresolved injury or illness causing the pain. In such a case, treatment of the underlying pathology could result in mitigation of pain [47].
Therapeutic intervention for Patient B begins with the use of NSAIDs and COX-2 inhibitors, especially for a pathology such as osteoarthritis, which is quite common among the elderly. As part of the assessment in this example, remember that elderly patients tend to have less total body water, decreased muscle tone, increased fat stores, and normal age-related degeneration of the liver and kidneys [47]. Unless there are other factors (e.g., current opioid use disorder), the best approach is to start low and titrate slow—use the smallest dose possible and increase it incrementally in small doses. The elderly often experience depression as part of their chronic pain; this should not be surprising, as living with unresolved pain each day can be psychologically taxing. Antidepressant agents, such as an SNRI, may be added to the care plan, with the caveat that there is an increased risk of falling [48]. Gabapentinoids may replace the antidepressant if the pain is neuropathic in nature, and opioids can be added on an as needed basis, though it is crucial to start at the low end of the dosing scale [48]. This follows the WHO guidance of NSAIDs first and opioids last.
While it is fine to conduct mental exercises with imaginary patients, the guiding standard for the clinician is whether the analgesia works. This is an important question, so at this point a small number of studies will be presented for your review.
In this example, Patient C, a man 71 years of age, presents with a severe case of recurring right sciatic pain [74]. On history and examination, the patient describes persistent pain at a scale of 8 out of 10, starting in the lumbar area of his back and running down his right thigh. Further comorbidities include chronic obstructive pulmonary disease (COPD) and previous right-side neck surgery to remove a buccal tumor. An MRI is ordered, revealing spondylolisthesis, which causes pain in lower back or legs at L5–S1, and the patient was also identified as having degenerative disk disease at L5–S1, and disk herniations at L3–L4, L4–L5, and L5–S1 [74]. This patient is taking 20 mg oxycodone daily in an effort to mitigate his pain.
The pain control team begins to titrate back the patient's oxycodone with tramadol and offers surgical decompression. After the patient refuses surgical intervention, the team decides to administer an ultrasound-guided caudal epidural steroid injection of triamcinolone 40 mg and 2% lidocaine 20 mg (local anesthetic) mixed in 12 mL of normal saline [74]. The mixture of normal saline is necessary because epidural injections require a greater volume to ensure the nerve roots are all bathed in the solution.
After the injection, Patient C's walking distance increases from 20 meters to 200 meters, and his pain score reduces from 10 to 7. One month later, the patient remains improved, but the team decides to add 5 mg oxycodone (25% of the original dose) as needed back to the patient's regimen. By his third month post procedure, the patient's pain score has dropped to 2. The multimodal plan, when compared with the singular large dose opioid plan, proved to be life-changing for this patient [74].
It is important to remember that pain is an adaptive mechanism to protect the body from comprise and prevent future involvement with the pain-generating stimulus. Modern medicine has many options to identify causes of pain and to treat the underlying problem while providing relief from pain [27]. However, pain management is an elusive goal for many patients. In the 1990s and 2000s, in an effort to address this problem, opioids were prescribed more freely. Unfortunately, this corresponded with an increase in opioid misuse and use disorder. In order to both assure that patient pain is managed and reduce the risks associated with opioids, numerous types of analgesics and techniques can be carefully mixed to decrease side effects while optimizing pain control.
It is crucial for all practitioners to carefully evaluate patients who are in pain to determine the extent to which the pain can be repaired or relieved. All available tools should be explored to treat the causes of pain at the local peripheral levels, the spinal cord levels, and the brain processing level. Familiarity with the pathophysiology of pain can guide good pharmacologic decisions and restore the patient's quality of life.
1. Hamilton G, Baskett T. In the arms of Morpheus: the development of morphine for postoperative pain relief. Can J Anaesth. 2000;47(4):367-374.
4. Raffa RB, Pergolizzi JV Jr, LeQuang JA, et al. The fentanyl family: a distinguished medical history tainted by abuse. J Clin Pharm Ther. 2018;43:154-158.
5. Hall JE, Hall ME. Guyton and Hall Textbook of Medical Physiology. 15th ed. Philadelphia, PA: Elsevier; 2026.
6. Leunig M, Beck M, Stauffer E, Hertel R, Ganz R. Free nerve endings in the ligamentum capitis femoris. Acta Orthop Scand. 2000;71:452-454.
8. Dubin AE, Patapoutian A. Nociceptors: the sensors of the pain pathway. J Clin Invest. 2010;120:3760-3772.
9. Mense S. Functional anatomy of muscle: muscle, nociceptors and afferent fibers. In: Mense S, Gerwin RD (eds). Muscle Pain: Understanding the Mechanisms. Berlin: Springer-Verlag; 2010: 17-48.
10. Black PH. Stress and the inflammatory response: a review of neurogenic inflammation. Brain Behav Immun. 2002;16:622-653.
11. Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain.Anes. 2018;129:343-366.
12. Yam MF, Loh YC, Tan CS, Khadijah Adam S, Abdul Manan N, Basir R. General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int J Mol Sci. 2018;19(8):21-64.
13. Petho G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev. 2012;92:1699775.
15. Kosse A, Kang H. Antiepileptic agents. In: Kaye A, Kaye A, Urman R (eds). Essentials of Pharmacology for Anesthesia, Pain Medicine and Critical Care. New York, NY: Springer; 2015.
17. Huffman J, Haas RE. The safe and effective use of pharmacological agents used for sedation during radiological procedures. J Radiol Nurs. 2014;33:132-144.
18. Abdallah CG, Salas R, Jackowski A, Baldwin P, Sato JR, Mathew SJ. Hippocampal volume and the rapid antidepressant effect of ketamine. J Psychopharmacol. 2015;29:591-595.
19. Fasick V, Spengler RN, Samankan S, Nader ND, Ignatowski TA. The hippocampus and TNF: common links between chronic pain and depression. Neurosci Biobehav Rev. 2015;53:139-159.
21. Ong WY, Stohler CS, Herr DR. Role of the prefrontal cortex in pain processing. Mol Neurobiol. 2019;56:1137-1166.
22. Aizenberg M, Rolon-Martinez S, Pham T, Rao W, Haas JS, Geffen MN. Projection from the amygdala to the thalamic reticular nucleus amplifies cortical sound responses. Cell Rep. 2019;28:605-615.
23. Luscher C, Malenka RC. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol. 2012;4(6):a005710.
24. Jensen MP, Brownstone RM. Mechanisms of spinal cord stimulation for the treatment of pain: still in the dark after 50 years. Eur J Pain. 2019;23:652-659.
25. Reus GZ, Abelaira HM, Tuon T, et al. Glutamatergic NMDA receptor as therapeutic target for depression. Adv Protein Chem Struct Biol. 2016;103:169-202.
27. Nguyen J, Haas R, Pugh L. The application of the theory of unpleasant symptoms to the education and practice of nurse anesthetists. NHIJ. 2017;1:000120.
28. Ma RSY, Kayani K, Whyte-Oshodi D, et al. Voltage-gated sodium channels as therapeutic targets for chronic pain. J Pain Res. 2019;12:2709-2722.
29. von Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73(4):638-652.
30. Widerström-Noga EG, Finnerup NB, Siddall PJ. Biopsychosocial perspective on a mechanisms-based approach to assessment and treatment of pain following spinal cord injury. J Rehabil Res Dev. 2009;46(1):1-12.
31. Institute of Medicine. Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research. Washington, DC: The National Academies Press; 2011.
32. Woolf CJ. Pain: moving from symptom control toward mechanism-specific pharmacologic management. Ann Intern Med. 2004;140:441-451.
33. American Society of Anesthesiologists. Practice guidelines for chronic pain management. Anesthesiol. 2010;112(4):810-833.
34. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895-926.
35. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1-32.
36. Chou R, Qaseem A, Snow V, et al. Diagnosis and treatment of low back pain: a joint clinical practice guideline from the American College of Physicians and the American Pain Society. Ann Intern Med. 2007;147(7):478-491.
37. Centers for Disease Control and Prevention. CDC clinical practice guideline for prescribing opioids for pain — United States, 2022. MMWR. 2022;71(3):1-95.
38. Cook CE, Rhon DI, Bialosky J, et al. Developing manual therapy frameworks for dedicated pain mechanisms. JOSPT Open. 2023;1(1):1-108.
39. Chimenti RL, Frey-Law LA, Sluka KA. A mechanism-based approach to physical therapist management of pain. Phys Ther. 2018;98(5):302-314.
40. Dworkin RH, O'Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237-251.
44. Yaksh TL, Wallace MS. Opioids, analgesia, and pain management. In: Brunton LL, Chabner BA, Knollmann BC (eds). Goodman and Gilman's The Pharmacological Basis of Therapeutics. 13th ed. New York, NY: McGraw-Hill; 2017: 481-525.
45. Pasternak GW. Opioid pharmacotherapy: from receptor to bedside. In: Inturrisi CE, Nicholson B, Pasternak GW (eds). Dual Opioid Therapy. London: The Royal Society of Medicine Press Limited; 2009.
46. Ghelardini C, Di Cesare Mannelli L, Bianchi E. The pharmacological basis of opioids. Clin Cases Miner Bone Metab. 2015;12(3):219-221.
47. Luchting B, Azad SC. Pain therapy for the elderly patient: is opioid-free an option? Curr Opin Anaesthesiol. 2019;32:86-91.
48. Schwan J, Sclafani J, Tawfik VL. Chronic pain management in the elderly. Anesthesiol Clin. 2019;37:547-560.
49. McKeen MJ, Quraishi SA. Clinical review of intravenous opioids in acute care. J Anesthiol Clin Sci. 2013;2:1-11.
50. Trang T, Al-Hasani R, Salvemini D, Salter MW, Gutstein H, Cahill CM. Pain and poppies: the good, the bad, and the ugly of opioid analgesics. J Neurosci. 2015;35(41):13879-13888.
51. Sprouse-Blum AS, Smith G, Sugai D, Parsa FD. Understanding endorphins and their importance in pain management. Hawaii Med J. 2010;69(3):70-71.
52. Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham DJ, Lambert DG. Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effect profile. Br J Anaesth. 2009;103(1):38-49.
53. Lesniak A, Lipkowski AW. Opioid peptides in peripheral pain control. Acta Neurobiol Exp. 2011;71(1):129-138.
54. Yaksh TL. Pharmacology and mechanisms of opioid analgesic activity. Acta Anaesthesiol Scand. 1997;41(1 Pt 2):94-111.
55. Atkinson TJ, Fudin J, Pandula A, Mirza M. Medication pain management in the elderly: unique and underutilized analgesic treatment options. Clin Ther. 2013;35(11):1669-1689.
56. Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician. 2008;11(2 Suppl):S133-S153.
57. Sindt J, Jenkinson R. Nonintravenous Opioids. In: Hemmings HC Jr, Egan T (eds). Pharmacology and Physiology for Anesthesia. Philadelphia, PA: Elsevier; 2019: 354-368.
58. LexiDrug. Available at https://online.lexi.com. Last accessed September 15, 2025.
59. Vallejo R, Barkin RL, Wang VC. Pharmacology of opioids in the treatment of chronic pain syndromes. Pain Physician. 2011;14(4):E343-E360.
60. Eisenberg E, McNicol ED, Carr DB. Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin. JAMA. 2005;293(24):3043-3052.
62. Sehgal N, Smith H, Manchikanti L. Peripherally acting opioids and clinical implications for pain control. Pain Physician. 2011;14(3):249-258.
63. Haeseler G, Foadi N, Ahrens J, Dengler R, Hecker H, Leuwer M. Tramadol, fentanyl and sufentanil but not morphine block voltage-operated sodium channels. Pain. 2006;126(1-3):234-244.
64. Armstrong SC, Cozza KL. Pharmacokinetic drug interactions of morphine, codeine, and their derivatives: theory and clinical reality, Part II. Psychosom. 2003;44(6):515-520.
67. Stavitskaya L, Coop A. Most recent developments and modifications of 14-alkylamino and 14-alkoxy-4,5-epoxymorphinan derivatives. Mini Rev Med Chem. 2011;11(12):1002-1008.
68. Hamann S, Sloan PA, Witt W. Low-dose intrathecal naloxone to enhance intrathecal morphine analgesia: a case report. J Opioid Manag. 2008;4(4):251-254.
69. Arbuck D, Gharibo C, Labhsetwar S, et al. Management of opioid tolerability and related adverse effects. J Medicine. 2010;3(1): 1-10.
70. Cruciani RA, Lussier D, Miller-Saultz D, Arbuck DM. Ultra-low dose oral naltrexone decreases side effects and potentiates the effect of methadone. J Pain Symptom Manage. 2003;25(6):491-494.
71. Raffa RB, Pergolizzi JV Jr, Tallarida RJ. The determination and application of fixed-dose analgesic combinations for treating multimodal pain. J Pain. 2010;11(8):701-709.
72. Scheiman JM. The use of proton pump inhibitors in treating and preventing NSAID-induced mucosal damage. Arthritis Res Ther. 2013;15(Suppl 3):S5.
73. Whittle SL, Colebatch AN, Buchbinder R, et al. Multinational evidence-based recommendations for pain management by pharmacotherapy in inflammatory arthritis: integrating systematic literature research and expert opinion of a broad panel of rheumatologists in the 3e Initiative. Rheumatology. 2012;51:1416-1425.
74. Gau P, Chen Y, Lo Y, Lu I, Chen P. Opioid-sparing multimodal analgesia for recurrent sciatic pain. Taiwan Journal of Pain. 2018;28:46-51.
75. Bekhit M, Navab K, Ghobrial A, Aust T. Nonopiod analgesic and adjunct drugs. In: Kaye A, Kaye A, Urman R (eds). Essentials of Pharmacology for Anesthesia, Pain Medicine and Critical Care. New York, NY: Springer; 2015: 148-156.
76. Smith HS, Kara DK, Argoff CE. Management of neuropathic pain: current insights and future perspectives. US Neurology. 2012;8(1):57-61.
77. Kulkantrakorn K. Emerging concepts and treatment in neuropathic pain. Neurology Asia. 2012;17:265-271.
79. European Urological Association. Guidelines on Pain Management and Palliative Care. Available at https://d56bochluxqnz.cloudfront.net/media/Pocket-Pain-Management-Urology_LR.pdf. Last accessed October 1, 2025.
80. Paul J, RArya A, Hurlburt L, et al. Femoral nerve block improves analgesia outcomes after total knoee arthroplasty. Anesth. 2010;113:1144-1162.
81. El-Boghdadly K, Pawa A, Chin KJ. Local anesthetic systemic toxicity: current perspectives. Local Reg Anesth. 2018;11:35-44.
82. Suzuki S, Gerner P, Lirk P. Local anesthetics. In: Hemmings HC Jr, Egan T (eds). Pharmacology and Physiology for Anesthesia. Philadelphia, PA: Elsevier; 2019.
84. Indiana Health Alert Network. Maximum recommended doses and duration of local anesthetics. In: Protocols IHaN. Iowa City, IA: University of Iowa Press; 2019.
85. Ciechanowicz S, Patil V. Lipid emulsion for local anesthetic systemic toxicity. Anesthesiology Research and Practice. 2012;2012:131784.
86. Ghai A, Gupta M, Hooda S, Singla D, Wadhera R. A randomized controlled trial to compare pregabalin with gabapentin for postoperative pain in abdominal hysterectomy. Saudi J Anaesth. 2011;5:252-257.
87. Imani F, Rahimzadeh P. Gabapentinoids: gabapentin and pregabalin for postoperative pain management. Anesth Pain Med. 2012;2:52-53.
88. Cheng TH. Spinal cord mechanisms of chronic pain and clinical implications. Curr Pain Headache Rep. 2010;14(3):213-220.
89. Xu B, Descalzi G, Ye HR, Zhuo M, Wang YW. Translational investigation and treatment of neuropathic pain. Mol Pain. 2012;8:15.
90. Hayashida KI, Obata H. Strategies to treat chronic pain and strengthen impaired descending noradrenergic inhibitory system. Int J Mol Sci. 2019;20(4):822.
91. Hoffman J. The historical shift in the perception of opiates: from medicine to social menace. JOPD. 1990;22:53-62.
92. Barkin RL, Barkin SJ, Barkin DS. Pharmacotherapeutic management of pain with a focus directed at the geriatric patient. Rheum Dis Clin North Am. 2007;33(1):1-31.
93. Yabeck-Karam V, Aouad M. Perioperative use of dexmedetomidine. Middle East J Anesthesiol. 2006;18:1043.
94. Seymore RJ, Manis MM, Coyne PJ. Dexmedetomidine use in a case of severe cancer pain. J Pain Palliat Care Pharmacother. 2019;33:34-41.
95. Brown E, Lydic R, Schiff N. General anesthesia, sleep and coma. N Engl J Med. 2010;363:2638-2650.
96. Calvey N, Williams N. Inhalational anesthesia agents. In: Calvey N, Williams N (ed). Principles and Practice of Pharmacology for Anaesthestists. Malden, MA: Blackwell Science; 2008: 129-148.
97. Calvey N, Williams N. Intravenous anaesthetic agents. In: Calvey N, Williams N (ed). Principles and Practice of Pharmacology for Anaesthestists. Malden, MA: Blackwell Science; 2008: 110-128.
98. Blonk MI, Koder BG, van den Bemt P, Huygen FJ. Use of oral ketamine in chronic pain management: a review. Eur J Pain. 2009;14(5):466-472.
99. Schwartzman RJ, Alexander GM, Grothusen JR. The use of ketamine in complex regional pain syndrome: possible mechanisms.Expert Rev Neurother. 2011;11(5):719-734.
100. Patil S, Anitescu M. Efficacy of outpatient ketamine infusions in refractory chronic pain syndromes: a 5-year retrospective analysis.Pain Med. 2012;13(2):263-269.
101. Schwartzman RJ, Alexander GM, Grothusen JR, Paylor T, Reichenberger E, Perreault M. Outpatient intravenous ketamine for the treatment of complex regional pain syndrome: a double-blind placebo-controlled study. Pain. 2009;147(1-3):107-115.
102. Quibell R, Prommer EE, Mihalyo M, Twycross R, Wilcock A. Ketamine. J Pain Symptom Manage. 2011;41:640-649.
103. Chen L, Malek T. Follow me down the K-hole: ketamine and its modern applications. Crit Care Nurs Q. 2015;38:211-216.
104. Cleveland Clinic. Date rape drugs: what parents should know. Cleve Clin J Med. 2001;68(6):551-552.
105. Abdallah CG, Sanacora G, Duman RS, Krystal JH. The neurobiology of depression, ketamine and rapid-acting antidepressants: is it glutamate inhibition or activation? Pharmacol Ther. 2018;190:148-158.
106. Xiong J, Lipsitz O, Chen-Li D et al. The acute antisuicidal effects of single-dose intravenous ketamine and intranasal esketamine in individuals with major depression and bipolar disorders: a systematic review and meta-analysis. J Psychiatr Res. 2021;134:57-68.
107. Silverman M, Hexem J. Nitrous oxide/oxygen sedation and the single-dose sedative. Inside Dentistry. 2011;42-54.
108. Whizar-Lugo VM, Heredia-Mota C, Camacho A. Back to the future: ketamine and nitrous oxide in major refractory depression.J Anesth Crit Care. 2017;9(3):1-5.
109. Vallejo MC, Zakowski MI. Pro-con debate: nitrous oxide for labor analgesia. Biomed Res Int. 2019;2019:4618798.
110. Faddy SC, Garlick SR. A systematic review of the safety of analgesia with 50% nitrous oxide: can lay responders use analgesic gases in the prehospital setting? Emerg Med J. 2005;22:901-908.
111. Urits I, Peck J, Orhurhu MS, et al. Off-label antidepressant use for treatment and management of chronic pain: evolving understanding and comprehensive review. Curr Pain Headache Rep. 2019;23:66.
112. Verdu B, Decosted I, Buclin T, Stiefel F, Berney A. Antidepressants for the treatment of chronic pain. Drugs. 2008;68:2611-2632.
113. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1-32.
114. Rahman W, D'Mello R, Dickenson AH. Peripheral nerve injury-induced changes in spinal alpha(2)-adrenoceptor-mediated modulation of mechanically evoked dorsal horn neuronal responses. J Pain. 2008;9:350-359.
115. Marks DM, Shah MJ, Patkar AA, et al. Serotonin-norepinephrine reuptake inhibitors for pain control: premise and promise. Cur Neuropharmacology. 2009;7(4):331-336.
116. Jackson K, St. Onge EL. Antidepressant pharmacotherapy: considerations for the pain clinician. Pain Practice. 2003;3:135-143.
117. Chan H, Fam J, Ng B. Use of antidepressants in the treatment of chronic pain. Annals Academy of Medicine Singapore. 2009;38:974.
118. World Health Organization. WHO Guidelines for the pharmacological and radiotherapeutic management of cancer pain in adults and adolescents. Geneva: World Health Organization; 2019.
119. Pollak A, Barnes L, Ciotola J, Gulli B. Emergency Care and Transportation of the Sick and Injured. Burlington, MA: Jones and Bartlett Learning; 2011.
120. McLaughlin DC, Cheah JW, Aleshi P, Zhang AL, Ma CB, Feeley BT. Multimodal analgesia decreases opioid consumption after shoulder arthroplasty: a prospective cohort study. J Shoulder Elbow Surg. 2018;27:686-691.
121. Guo JG, Zhao LP, Rao YF et al. Novel multimodal analgesia regimen improves post-TACE pain in patients with hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2018;17:510-516.
1. Dowell D, Ragan KR, Jones CM, Baldwin GT, Chou R. CDC clinical practice guideline for prescribing opioids for pain—United States, 2022. MMWR. 2022;71(RR3):1-95. Available at https://www.cdc.gov/mmwr/volumes/71/rr/rr7103a1.htm. Last accessed October 23, 2025.
2. Schwenk ES, Viscusi ER, Buvanendran A, et al. Consensus guidelines on the use of intravenous ketamine infusions for acute pain management from the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg Anesth Pain Med. 2018;43(5):456-466. Available at https://rapm.bmj.com/content/rapm/43/5/456.full.pdf. Last accessed October 23, 2025.
3. Hsu J, Mir H, Wally M, Seymour R, the Orthopaedic Trauma Association Musculoskeletal Pain Task Force. Clinical practice guidelines for pain management in acute musculoskeletal injury. J Orth Trauma. 2019;33(5):e158-e182. Available at https://journals.lww.com/jorthotrauma/fulltext/2019/05000/clinical_practice_guidelines_for_pain_management.11.aspx. Last accessed October 23, 2025.
Mention of commercial products does not indicate endorsement.