The U.S. Food and Drug Administration (FDA) regulates many products, but despite the agency's expansive reach, many healthcare professionals may be unfamiliar with what the FDA regulates and how it determines whether a drug is approved, as well as the processes and standards for drug approval. This course aims to educate physicians and other healthcare professionals who prescribe drugs on how the FDA ensures the safety, efficacy, and security of these products. Participants will learn the regulatory considerations for how clinical trials are designed and conducted, as well as the general criteria for how new drugs are developed, tested, approved, and monitored. This course covers only products regulated by the FDA's Center for Drug Evaluation and Research (CDER).
- INTRODUCTION
- PRESCRIBER KNOWLEDGE OF DRUG APPROVAL STANDARDS
- CENTER FOR DRUG EVALUATION AND RESEARCH
- DEFINING SAFE AND EFFECTIVE
- THE DRUG DEVELOPMENT AND APPROVAL PROCESS
- EXPEDITED REVIEW PROGRAMS
- POST-APPROVAL RESPONSIBILITIES
- RARE DISEASES DRUG DEVELOPMENT
- EXPANDED ACCESS
- FURTHER RESOURCES
- CONCLUSION
- Works Cited
This course is designed for all physicians, pharmacy professionals, and nurses involved prescribing, dispensing, and/or administering medications.
The purpose of this course is to educate physicians and other healthcare professionals who prescribe drugs on how the FDA ensures the safety, efficacy, and security of approved products.
Upon completion of this course, you should be able to:
- Explain the U.S. Food and Drug Administration's (FDA's) role in drug regulation.
- Describe the origin of safety and efficacy standards.
- Outline the steps of drug development and approval.
- Differentiate the FDA's expedited new drug review pathways.
- Describe the post-approval responsibilities of the FDA, manufacturers, and prescribers.
- Explain expanded access, rare disease development, and orphan drug development.
John J. Whyte, MD, MPH, is currently the Chief Medical Officer at WebMD. In this role, he leads efforts to develop and expand strategic partnerships that create meaningful change around important and timely public health issues. Previously, Dr. Whyte was the Director of Professional Affairs and Stakeholder Engagement at the FDA’s Center for Drug Evaluation and Research and the Chief Medical Expert and Vice President, Health and Medical Education at Discovery Channel, part of the media conglomerate Discovery Communications.
Prior to this, Dr. Whyte was in the Immediate Office of the Director at the Agency for Healthcare Research Quality. He served as Medical Advisor/Director of the Council on Private Sector Initiatives to Improve the Safety, Security, and Quality of Healthcare. Prior to this assignment, Dr. Whyte was the Acting Director, Division of Medical Items and Devices in the Coverage and Analysis Group in the Centers for Medicare & Medicaid Services (CMS). CMS is the federal agency responsible for administering the Medicare and Medicaid programs. In his role at CMS, Dr.Whyte made recommendations as to whether or not the Medicare program should pay for certain procedures, equipment, or services. His division was responsible for durable medical equipment, orthotics/prosthetics, drugs/biologics/therapeutics, medical items, laboratory tests, and non-implantable devices. As Division Director as well as Medical Officer/Senior Advisor, Dr. Whyte was responsible for more national coverage decisions than any other CMS staff.
Dr. Whyte is a board-certified internist. He completed an internal medicine residency at Duke University Medical Center as well as earned a Master’s of Public Health (MPH) in Health Policy and Management at Harvard University School of Public Health. Prior to arriving in Washington, Dr. Whyte was a health services research fellow at Stanford and attending physician in the Department of Medicine. He has written extensively in the medical and lay press on health policy issues.
Junyang Wang, MSc, is a Clinical Analyst in Professional Affairs and Stakeholder Engagement at the Center for Drug Evaluation and Research at the U.S. Food and Drug Administration. In this role, Mr. Wang works closely on issues of demographic subgroup analyses, variability to drug response, and the Drug Trials Snapshots transparency initiative. He has led large data science projects combining patient demographic data with clinical trial site geographic data to better understand the global make-up of clinical trials across the world. Previously, Mr. Wang worked for the Public Relations firm Edelman in Los Angeles, conducted health economic research at the National University of Singapore, and assisted in developing the new Duke-Kunshan University in Kunshan, China. He completed his Masters of Science in Global Health as well as a Bachelor’s of Science in Psychology from Duke University.
Noah Goetzel, is a Master of Public Health (MPH) candidate at the Columbia University Mailman School of Public in the Health Promotion Research and Practice certificate program and earned his Bachelor of Arts degree in Journalism and Radio, TV, and Film Studies at the University of Wisconsin-Madison. He is studying the intersection between health behavior change theory and communication campaign planning in the digital age. His thesis project evaluates the effectiveness of the Health Mentorship program at EHE Health in helping patients achieve their weight loss, nutrition, physical activity, stress management, sleep, and substance use goals. As a creative intern at EHE Health, Mr. Goetzel leads a study measuring the impact of wearable posture trainer biofeedback devices on musculoskeletal symptoms, manages EHE Health’s social media accounts, and leads patient health incentive implementation efforts. Prior to his internship, Mr. Goetzel was a liaison between clinical drug reviewers and patient and professional group stakeholders as an ORISE Fellow at the U.S. Food and Drug Administration (FDA).
Contributing faculty, John J. Whyte, MD, MPH, has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Contributing faculty, Junyang Wang, MSc, has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Contributing faculty, Noah Goetzel, has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
John M. Leonard, MD
Mary Franks, MSN, APRN, FNP-C
Randall L. Allen, PharmD
The division planners have disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
Sarah Campbell
The Director of Development and Academic Affairs has disclosed no relevant financial relationship with any product manufacturer or service provider mentioned.
The purpose of NetCE is to provide challenging curricula to assist healthcare professionals to raise their levels of expertise while fulfilling their continuing education requirements, thereby improving the quality of healthcare.
Our contributing faculty members have taken care to ensure that the information and recommendations are accurate and compatible with the standards generally accepted at the time of publication. The publisher disclaims any liability, loss or damage incurred as a consequence, directly or indirectly, of the use and application of any of the contents. Participants are cautioned about the potential risk of using limited knowledge when integrating new techniques into practice.
It is the policy of NetCE not to accept commercial support. Furthermore, commercial interests are prohibited from distributing or providing access to this activity to learners.
Supported browsers for Windows include Microsoft Internet Explorer 9.0 and up, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Supported browsers for Macintosh include Safari, Mozilla Firefox 3.0 and up, Opera 9.0 and up, and Google Chrome. Other operating systems and browsers that include complete implementations of ECMAScript edition 3 and CSS 2.0 may work, but are not supported. Supported browsers must utilize the TLS encryption protocol v1.1 or v1.2 in order to connect to pages that require a secured HTTPS connection. TLS v1.0 is not supported.
The role of implicit biases on healthcare outcomes has become a concern, as there is some evidence that implicit biases contribute to health disparities, professionals' attitudes toward and interactions with patients, quality of care, diagnoses, and treatment decisions. This may produce differences in help-seeking, diagnoses, and ultimately treatments and interventions. Implicit biases may also unwittingly produce professional behaviors, attitudes, and interactions that reduce patients' trust and comfort with their provider, leading to earlier termination of visits and/or reduced adherence and follow-up. Disadvantaged groups are marginalized in the healthcare system and vulnerable on multiple levels; health professionals' implicit biases can further exacerbate these existing disadvantages.
Interventions or strategies designed to reduce implicit bias may be categorized as change-based or control-based. Change-based interventions focus on reducing or changing cognitive associations underlying implicit biases. These interventions might include challenging stereotypes. Conversely, control-based interventions involve reducing the effects of the implicit bias on the individual's behaviors. These strategies include increasing awareness of biased thoughts and responses. The two types of interventions are not mutually exclusive and may be used synergistically.
#95001: Expanding the Options: The Drug-Approval Process in the United States
Products regulated by the U.S. Food and Drug Administration (FDA) comprise about one-fifth of every dollar spent on consumer products in the United States [1]. These products also account for approximately 15% of all U.S. imports and 15% of exports [1]. FDA-regulated products originate from more than 150 countries and nearly 275,000 facilities, of which 144,606 are overseas. An excess of $2.4 trillion is spent annually on these goods, including drugs, biologics, foods, dietary supplements, medical devices, cosmetics, veterinary products, tobacco products, and electronic products that emit radiation. In general, the FDA is responsible for protecting the public health by ensuring the safety, efficacy, and security of human and veterinary drugs, biologic products, and medical devices [2]. The administration also ensures the safety of the nation's food supply, cosmetics, and products that emit radiation. It achieves this by enforcing Title 21 of the Code of Federal Regulations, which governs food and drugs within the United States.
Prescription drugs have become a regular part of life for a significant portion of the U.S. population. Nearly half of Americans (45.4%) have taken at least one prescription drug in the past 30 days, and 11% of the population has taken five or more in that span [3]. Altogether, about 3.8 billion prescriptions for approximately 17,000 medical drug products were filled at retail pharmacies nationwide in 2019 [4]. One of the FDA's primary roles in protecting the public's health is determining whether these drugs are safe and effective to use.
For a drug to be approved, the sponsor must provide substantial evidence of effectiveness, and the benefits must outweigh the risks. The Center for Drug Evaluation and Research (CDER) oversees the drug development process by reviewing product safety and efficacy. Pharmaceutical companies and other investigators, collectively known as sponsors, perform a series of preclinical (animal) and clinical (human) trials, which the FDA monitors, to test if the drug is effective and safe. If CDER's review establishes that the drug's benefits outweigh its known risks for the proposed use, the drug is approved for sale.
Even after the drug is on the market, the FDA continues to monitor its performance in several ways. One of those ways is through MedWatch, the agency's safety information and adverse event reporting program, which receives reports of suspected adverse events (potential side effects) from consumers, healthcare practitioners, and pharmaceutical companies. If an unexpected drug-related health risk is detected, the FDA may alert consumers and healthcare professionals through a variety of mechanisms, such as the Drug Safety Communication. The FDA continually evaluates a new drug and revisits these questions continually over the drug's lifecycle.
This course aims to educate physicians and other healthcare professionals about CDER's rigorous and complex drug review process. The key question during the drug-review process is to determine if a new drug is safe and effective for its intended use. That calculated risk-benefit analysis will indicate whether the drug can earn FDA's approval to enter the U.S. market.
A 2016 national survey of board-certified internists and specialists revealed substantial deficits in knowledge of the meaning of FDA approval [5]. According to the survey, only 1% of physicians who were asked a series of survey questions about approval standards answered all three questions correctly. The surveys conducted on a random sample of active board-certified physicians showed the following significant misunderstandings [5]:
73% of respondents incorrectly believed FDA approval typically means that a drug is as effective as other drugs approved to treat the same condition.
70% incorrectly believed drug approval required a statistically significant and clinically important effect.
79% of respondents were either "a little familiar" or "not familiar at all" about the breakthrough therapy designation for drugs.
77% falsely believed that when the FDA calls a drug a "breakthrough" it has high-quality evidence showing it is safer than a currently approved treatment.
64% of physicians were incorrectly "fairly certain" or "very certain" that breakthrough drugs represented a "major advance" over currently approved treatments for the same indication.
94% preferred using a hypothetical FDA-designated breakthrough drug, compared with a drug that was not considered breakthrough drug but still met the FDA's definition of the breakthrough designation of having "early promising study results but not having been shown to improve survival or disease-related symptoms."
As the largest of the FDA's centers, CDER's nearly 4,500 employees protect and promote the health of Americans by making sure safe and effective drugs are available. CDER promotes the safe use of these drugs and helps ensure that drugs meet established quality standards. These drugs include prescription and over-the-counter medications along with generic drugs and some biologic therapeutics. CDER faces a multitude of challenges due to the complexity of the human drug supply and drug development pipeline.
From aspirin to cancer treatments, CDER has oversight responsibilities for prescription, over-the-counter, and generic drugs. The products differ in significant ways:
Prescription drugs: Any drug products that require a prescriber's authorization to purchase
Generic drugs: A drug product that is equivalent to brand name products in terms of quality and performance
Over-the-counter drugs: Products available to consumers without a physician's prescription
Drug sponsors produce and test drugs for CDER to review. The sponsor must first submit a drug's proposed labeling and evidence demonstrating the product's safety and efficacy for a specific disease or condition. A team of CDER physicians, statisticians, chemists, pharmacologists, and other scientists reviews the new drug application (NDA) [6]. The drug can be approved if the review team determines the drug's benefits outweigh its known risks and that the drug can be manufactured in a way that ensures a quality product [7].
After CDER grants approval for a new drug to be marketed in the United States, the Center conducts post-marketing surveillance. It is not possible to predict all adverse effects of a drug based on clinical trials of limited duration using a limited population during drug development, so the FDA maintains a system of post-marketing surveillance and risk assessment programs to identify and monitor adverse events that did not appear during the drug-approval process.
As discussed, drugs intended for human use are evaluated by CDER to ensure that drugs marketed in the United States are safe and effective. Although the FDA is recognized today as one of the world's foremost institutional authorities for reviewing and evaluating controlled clinical drug trials, the established standards of safe and effective drugs took decades to evolve into what they are today.
Before President Roosevelt signed the Federal Food, Drug, and Cosmetic (FD&C) Act of 1938 into law, manufacturers did not have to show that a drug was safe before it could be marketed. There were no scientific requirements for testing or approval nor were there any data submission requirements for companies marketing drugs. The FD&C Act of 1938 established the "safety" standards still in use today and subjected new drugs to premarket safety evaluation for the first time with two key changes:
Premarket notification to the FDA: Any new drug must have submitted an NDA, which came into effect (essentially approved) after six months of submission, unless objected by the FDA.
Demonstration of safety: The FDA could refuse to approve the NDA due to safety or labeling issues.
Although the law did not specify the kinds of tests that were required for approval, the new authority allowed drug officials to formally block the marketing of a new drug or delay it by requiring additional data. Similarly, applications could be refused if test results did not show that the drug was safe or if the labeling was false or misleading for the proposed indication. The Act also gave regulators limited powers of negotiation over scientific study and approval requirements with the pharmaceutical industry and the medical profession [8].
No drug is absolutely safe, as all drugs have side effects. Therefore, safety is evaluated along with the benefits of the drug and how they appear to outweigh the known risks. Although there is no strict definition of how to evaluate a drug's risk versus benefit, the FDA requires an integrated summary of safety as well as updates of safety information within the Code of Federal Regulations (21CFR 314.50) [9].
The applicant must submit an integrated summary of all available information about the safety of the drug product, including pertinent animal data, demonstrated or potential adverse effects of the drug, clinically significant drug-drug interactions, and other safety considerations, such as data from epidemiologic studies of related drugs. The safety data must be presented by gender, age, and racial subgroups. When appropriate, safety data from other subgroups of the population of patients treated also must be presented, such as for patients with renal failure or patients with different levels of severity of the disease. A description of any statistical analyses performed in evaluating safety data should also be included [9].
The applicant must, under section 505(i) of the FD&C Act, periodically update its pending NDA with new safety information learned about the drug that may reasonably affect the statement of contraindications, warnings, precautions, and adverse reactions in the draft labeling and, if applicable, any medication guide. These "safety update reports" must include the same kinds of information (from clinical studies, animal studies, and other sources) and must be submitted in the same format as the integrated summary. In addition, the reports must include the case report forms for each patient who died during a clinical study or who did not complete the study because of an adverse event (unless this requirement is waived) [9].
The effectiveness requirement for drug approval was added to the FD&C Act in 1962 (as part of what is known as the Kefauver-Harris Amendments) following the worldwide thalidomide drug disaster of 1961. The 1962 Act made at least three significant changes [35]:
Instead of a six-month automatic NDA approval if there was no objection by the FDA, now the FDA had to give a positive approval before the drug could be marketed.
It established the meaningful requirement to study drugs under an investigational new drug (IND) application with explicit informed consent from the study participants.
Applications must demonstrate the drug had substantial evidence of efficacy.
These new amendments explicitly stated that the FDA would rely on scientific testing and that new drug approvals would be based not only upon proof of safety but also on "substantial evidence" of a drug's efficacy (i.e., the impact of a drug in a clinical trial setting). Substantial evidence was defined in section 505(d) of the Act as [10]:
…evidence consisting of adequate and well-controlled investigations, including clinical investigations, by experts qualified by scientific training and experience to evaluate the effectiveness of the drug involved, on the basis of which it could fairly and responsibly be concluded by such experts that the drug will have the effect it purports or is represented to have under the conditions of use prescribed, recommended, or suggested in the labeling or proposed labeling thereof.
Since 1962, the FDA has overseen substantial refinements to the broad legal requirement that post-1962 new drugs be approved on the basis of "adequate and well-controlled" studies [11]. With regard to quantity, it has been the FDA's position that Congress generally intended to require at least two adequate and well-controlled studies, each convincing on its own, to establish effectiveness. The FDA's position is based on the language in the statute and the legislative history of the 1962 amendments. Section 505(d) of the Act uses the plural form in defining "substantial evidence" as "adequate and well-controlled investigations, including clinical investigation." Section 505(b) of the Act, which lists the contents of an NDA, also uses the plural "investigations." Language in a Senate report suggested that the phrase "adequate and well-controlled investigations" was designed not only to describe the quality of the required data but also the "quantum" of required evidence [12]. The three main goals of "adequate and well-controlled investigations" are to ensure:
A valid control group, whereby the control group is very similar to the test group so one can isolate the difference due to drug given
Bias minimization of the clinical trial (in how test and control group are selected, treated, observed, assessed, and analyzed)
Documentation of sufficient study details to allow a critical evaluation of whether the characteristics of an adequate and well-controlled study are present
Nevertheless, the FDA has been flexible within the limits imposed by the congressional scheme, broadly interpreting the statutory requirements to the extent possible when the data on a particular drug were convincing. In other cases, the FDA has relied on only a single adequate and well-controlled efficacy study to support approval—generally only in cases in which a single multicenter study of excellent design provided highly reliable and statistically strong evidence of an important clinical benefit (e.g., an effect on survival) and a confirmatory study would have been difficult to conduct on ethical grounds [10].
After an NDA is filed, an FDA review team of medical doctors, chemists, statisticians, microbiologists, pharmacologists, and other experts evaluates whether the sponsor's studies show that the drug is safe and effective for its proposed use. This often requires demonstration that the benefits of the drug outweigh its risks for the patient population for which the drug is indicated. If the FDA decides that the benefits of a drug outweigh the known risks, the review team will approve of the drug and it can be marketed in the United States. But if there are problems with an NDA or if more information is necessary to make that determination, the FDA may issue a complete response letter [14]. A "complete response" indicates that FDA has completed its review and has found issues that must be addressed before the drug can be approved. A company has the option to re-submit their application for approval after addressing the issues cited by the FDA.
A drug's path from the lab to the pharmacy can take many years and undergo several iterations. Often, a drug is developed to treat a specific disease. However, an important use of a drug may occasionally be discovered by accident. Most drugs that undergo preclinical testing are never tested on humans or reviewed by the FDA. The drugs that advance to the clinical trial stage of drug development undergo the agency's rigorous evaluation process, which scrutinizes everything about the drug, from clinical trial design to the severity of side effects, to the conditions under which the drug is manufactured.
The development process for drugs involves five basic steps:
Drug discovery and development: Research for a new drug begins in the laboratory.
Preclinical research: Drugs undergo laboratory and animal testing to answer basic questions about safety.
Clinical research: Drugs are tested on people to make sure they are safe and effective.
FDA review: FDA teams thoroughly review all of the submitted data related to the drug and make a decision to approve or not to approve it.
FDA post-market safety monitoring: FDA monitors drug safety after products are available for use by the public.
New drugs shape the practice of medicine by offering patients novel therapies for a wide array of common and rare conditions. There are many avenues for discovering new drugs, but researchers typically begin this process through one of the following methods [15]:
Learning new information about a disease process that enables researchers to design a product that stops or reverses the effects of the disease
Conducting many tests of molecular compounds to find possible benefits against a variety of diseases
Finding unanticipated effects from existing treatments
Discovering new technologies, such as those that target medical products to specific sites within the body or manipulate genetic material
Thousands of compounds may be potential candidates for medical product development[15]. After early testing, however, only a small number of them remain promising enough for further study (Figure 1). After researchers have identified a promising compound for development, they conduct a series of experiments to learn about:
How the molecular compound is absorbed, distributed, metabolized, and excreted
Potential benefits and mechanisms of action
Optimal dosage
The best way to deliver the drug (e.g., orally or by injection)
Adverse effects (i.e., toxicity)
How the compound affects different demographics of people
How it interacts with other drugs and treatments
Effectiveness compared with similar drugs
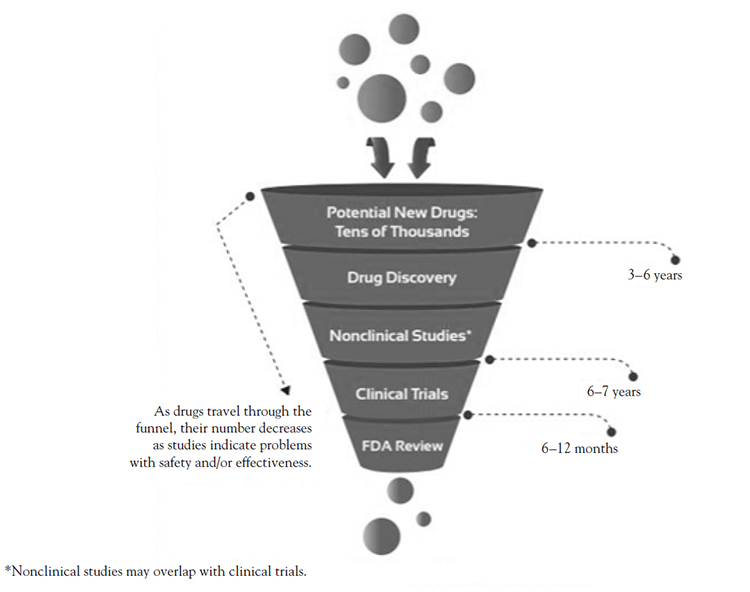
During a new drug's early development process, the sponsor's primary goal is to determine if the drug is reasonably safe for human use and if its course of action, or pharmacologic activity, justifies commercial development. Preclinical studies are typically not very large but must provide detailed information on dosing and toxicity levels of IND products. The sponsor has three options for fulfilling this requirement [16]:
Compiling existing nonclinical data on past laboratory or animal studies on the compound
Compiling data from previous clinical testing or marketing of the drug in the United States or countries with similar populations
Conducting new preclinical studies designed to provide the necessary evidence to support the safety of administering the compound to humans
To find out whether a drug has the potential to cause serious harm or toxicity, the drug manufacturer must conduct in-vitro preclinical research or in-vivo research using non-human living organisms. Preclinical studies often involve both experiment types to provide detailed information on dosing and toxicity levels. Researchers are required to use good laboratory practices, which set basic requirements for study conduct, personnel, facilities, equipment, protocol, operating procedures, study reports, and quality assurance oversight.
After preclinical testing, the FDA generally asks sponsors to develop a pharmacologic profile of the drug, determine its acute toxicity in two or more animal species, and conduct short-term toxicity studies. Preclinical testing does not always end after the application is submitted and ruled safe to proceed with clinical trials. Additional nonclinical studies may be required to support the safety of clinical trials, as they last longer and include more human subjects. Preclinical studies are also necessary for determining the dose, schedule, and duration of administration for humans.
If the sponsor feels the evidence of preclinical testing supports the product's safety, they will submit an IND application to demonstrate the results of preclinical testing to the FDA. Usually, the company submits an IND application for FDA's review prior to testing in humans. An IND presents data from testing on laboratory animals to prove the drug will not expose human subjects to unreasonable risks during early-stage clinical trials. When a sponsor decides to test a new molecular compound's effect on humans, the molecule changes in legal status and becomes a new drug subject to the rules of the drug regulatory system. Sponsors cannot start clinical investigations until an IND is in effect.
INDs can fall into either a commercial or research (non-commercial) category. Commercial INDs are typically submitted by companies that hope to gain marketing approval for a new pharmaceutical product. There are three types of research INDs [17]:
Investigator IND: Submitted by a physician who initiates and conducts an investigation. The investigational drug is administered or dispensed under the physician's immediate direction. A physician might submit a research IND to propose studying an unapproved drug or to study an approved product for a new indication or in a new patient population.
Emergency use IND: Allows the FDA to authorize use of an experimental drug in an emergency situation that does not allow time for submission of an IND in accordance with federal regulations (21CFR, Sec. 312.23 or Sec. 312.20). It is also used for patients who do not meet the criteria of an existing study protocol or if no approved study protocol exists.
Treatment IND: Submitted for experimental drugs showing promise in clinical testing for serious or immediately life-threatening conditions while the final clinical work is conducted and FDA review occurs.
All IND applications must include:
Preclinical data from animal pharmacology and toxicity studies, including prior experience with the drug in humans, if possible
Manufacturing information ensuring the drug company can produce and supply consistent batches of the drug
Clinical protocols and investigator information, such as informed consent and institutional review board sign-off, detailing how clinical trials on humans would be designed to prevent exposure to unnecessary risks
After IND submission, the FDA has 30 days to review the application for safety to ensure human research participants will not be subjected to unreasonable risk. The agency may or may not contact the sponsor about its determination. Unless the FDA notifies the sponsor that a clinical hold has been placed to delay or stop investigations, clinical trials may begin 30 days after the FDA receives the IND.
While preclinical research answers basic questions about a drug's safety, it is not a substitute for clinical research studies—trials conducted on people to assess ways the drug will interact with the human body [18]. The goal of clinical trials is to determine whether an investigational drug is effective and what side effects it may cause. As developers design the clinical study, they consider what they want to accomplish for each of the different phases of clinical research.
Researchers design clinical trials to evaluate the safety and efficacy of a medical product. These trials follow a specific study plan (protocol) developed by the sponsor. Before a clinical trial begins, researchers review prior information about the drug to develop research questions and objectives, including:
Who qualifies to participate (selection criteria)
How many people will be part of the study during each phase of clinical trials
How long the study will last
Whether there will be a control group and other ways to limit research bias
How the drug will be given to patients and at what dosage
What assessments will be conducted, when, and what data will be collected
How the data will be reviewed and analyzed
Clinical trials follow a typical progression from early, small-scale Phase 1 studies to late-stage, large-scale Phase 3 studies. Before initiating a clinical trial, sponsors must conduct early-phase nonclinical studies and file and have an effective IND, meaning the IND was submitted to the FDA at least 30 days prior to the initiation of clinical trials without a clinical hold.
Phase 1
Study Participants: Generally, 20 to 100 healthy volunteers or patients with the disease/condition who could benefit from the product tested in the IND
Length of Study: Several months
Purpose: Safety and dosage
During Phase 1 studies, researchers typically test a new drug in normal (healthy) volunteers. However, some Phase 1 studies test new drugs in patients with a particular disease. For example, if a new drug is intended for use in patients with certain cancers, researchers conduct Phase 1 studies in patients with that type of cancer.
Phase 1 studies are closely monitored and gather information about how a drug interacts with the human body. Researchers adjust dosing schemes based on animal data to find out how much of a drug the body can tolerate and what its acute side effects are.
As a Phase 1 trial continues, researchers answer research questions related to how the drug works in the body, the side effects associated with increased dosage, and early information about its effectiveness to determine how best to administer the drug, limit risks, and maximize possible benefits. This is important to the design of Phase 2 studies.
Phase 2
Study Participants: Up to several hundred people with the disease/condition
Length of Study: Several months to two years
Purpose: Efficacy and side effects
Approximately 70% of drugs that reach Phase 1 move to Phase 2 [18]. Patients with the disease or condition that the investigational drug intends to treat comprise the subjects enrolled in Phase 2 studies. These studies typically involve a few hundred patients but are not large enough to show whether the drug will be beneficial. Instead, Phase 2 studies provide researchers with additional safety data. Researchers use these data to refine research questions, develop research methods, and design new Phase 3 research protocols. Approximately 33% of drugs that reach Phase 2 move to Phase 3 [4].
Phase 3
Study Participants: Typically, 300 to 3,000 volunteers who have the disease or condition
Length of Study: One to four years
Purpose: Efficacy and monitoring of adverse reactions
Researchers design Phase 3 studies to demonstrate whether or not a product offers a treatment benefit to a specific population. Sometimes known as pivotal studies, these studies involve 300 to 3,000 participants.
Phase 3 studies provide most of the safety data. In previous studies, it is possible that less common side effects might have gone undetected. Because these studies are larger and longer in duration, the results are more likely to show long-term or rare side effects. Approximately 25% to 30% of drugs that reach Phase 3 are approved and move to the post-marketing surveillance phase[4].
Clinical trials do not officially end after NDA approval. Safety and efficacy evaluations continue through the FDA's post-market safety monitoring. Several thousand volunteers who have the drug's indicated disease or condition are part of this final, ongoing phase of product evaluation. This stage is sometimes referred to as Phase 4 clinical trials.
As discussed, the NDA is the vehicle through which drug sponsors formally propose that the FDA approve a new drug, often called a new molecular entity. Data gathered during animal studies and human clinical trials, along with descriptions of manufacturing procedures and additional drug information, are part of the NDA. Overall, the NDA aims to provide the FDA review team with ample information needed to answer the following questions:
Is the drug safe and effective for its proposed use(s)?
Do the benefits of the drug outweigh the risk?
Is the drug's proposed labeling (package insert) appropriate and what information should that labeling contain?
Do the methods used in manufacturing and the controls used to maintain the drug's quality adequately preserve the drug's identity, strength, quality, and purity?
The NDA explains to reviewers what happened during the clinical tests, what the ingredients of the drug are, the results of the animal studies, how the drug behaves in the body, and how it is manufactured, processed, and packaged. Key NDA submission requirements:
Preclinical pharmacology and toxicology data
Human pharmacology and pharmacokinetics data (Phase 1)
Clinical data demonstrating efficacy (Phases 2 and 3)
Chemistry and manufacturing information providing assurance of drug identity, reproducibility, purity, quality, strength, and stability
After the drug developer has filed the NDA to the FDA for review, the CDER review team has 60 days to decide whether the application is complete. The FDA review team thoroughly examines all submitted data on the drug and makes a decision to accept the application for review. If the reviewers determine the application is complete, they have 6 to 10 months to evaluate whether to approve the drug.
Each member of the review team conducts a full review of his or her section of the application. For example, the medical officer and the statistician review clinical data, while a pharmacologist reviews the data from animal studies. There is also a supervisory review for each technical discipline within the team. CDER inspectors may travel to clinical study sites to conduct a routine inspection. The agency looks for evidence of fabrication, adulteration, manipulation, or withholding of data. Then, the project manager assembles all individual reviews and other documents, such as the inspection report, into an "action package." This document becomes the record for FDA review. The review team issues a recommendation, and a senior FDA official makes a decision.
In cases in which CDER determines that a drug has been shown to be safe and effective for its intended use, scientists proceed to work with the applicant to develop and refine prescribing information, also referred to as the labeling. Labeling accurately and objectively describes the basis for approval and how best to use the drug.
Remaining issues often need to be resolved before the drug can be approved for marketing. In some cases, CDER requires the drug developer to address questions based on existing data. In other cases, additional studies are required. At this point, the developer can decide whether or not to continue further development. If a developer disagrees with an FDA decision, there are mechanisms for formal appeal.
In the event that questions that require additional consideration arise during the NDA review process, the FDA may organize a meeting of one of its Advisory Committees to gain independent, expert advice and to allow the public to make comments. These Advisory Committees include technically qualified experts in their field (e.g., clinical medicine, engineering, biological sciences) as well as a patient representative who provides input from the patient perspective and may vote when serving on committees that review drug and biologic therapies. The most impactful contribution Advisory Committee members make is not their final vote; rather, it is their contribution to the discussion of a product's safety, effectiveness, or clinical development [19]. The Committee recommendations are not binding for FDA action, and as their name implies, their recommendations are advisory.
Along with the various steps involved in reviewing a new drug through the standard pathway, CDER also has the flexibility to approve drugs through various expedited programs. An increasing number of new drugs are moving through the FDA's approval process at a quicker pace. In fact, a 2015 British Medical Journal study found the number of approved drugs qualifying for FDA's expedited drug development and approval programs increased 2.6% annually between 1987 and 2014[20]. The following sections will help clarify the FDA's drug-approval standards, along with the breakthrough therapy designation and other frequently misunderstood aspects of FDA's regulatory authority.
Expedited review means the FDA can hasten its review process to help the product reach patients sooner if the new drug addresses an unmet medical need and treats a serious or life-threatening condition[21]. Expediting the availability of life-changing drugs benefits everyone. This is especially true if a drug becomes the first available treatment for a particular condition or has advantages over existing treatments[6]. In 2016, for example, CDER approved the first ever treatments for spinal muscular atrophy and Duchenne muscular dystrophy. An estimated 60% of novel drugs approved in 2019–2022 benefited from one of the FDA's expedited review programs (Table 1)[22,23]:
Accelerated approval
Fast track
Breakthrough therapy
Priority review
SUMMARY DESCRIPTIONS OF DRUGS ELIGIBLE FOR EXPEDITED REVIEW
| Expedited Program | Characteristics of Eligible Drugs | ||||
|---|---|---|---|---|---|
| Fast track |
| ||||
| Breakthrough therapy |
| ||||
| Accelerated approval | A drug that treats a serious medical condition and generally provides a
meaningful advantage over available therapies. The drug demonstrates an effect on:
| ||||
| Priority review |
|
It can take years to learn whether a drug truly impacts how a patient survives, feels, or functions [24]. For patients whose illness is potentially terminal, the length of time it typically takes for approval can be problematic. Accelerated approval allows drugs for serious conditions that fill unmet medical needs to be expedited during the review process and approved based on a surrogate or intermediate clinical endpoint [27].
For example, when acquired immunodeficiency syndrome (AIDS) became a leading cause of death among men 25 to 44 years of age in 1992, patients desperately needed access to therapies. Instead of evaluating whether an antiviral drug prolonged patient survival, the FDA reduced clinical trial time by using CD4 cell counts and measures of viral load as indicators for predicting increases in survival rates [25,26]. The FDA approved zidovudine based on these early indicators, known as surrogate or intermediate clinical biomarkers, and required the sponsor to confirm the predicted clinical benefits after approval. This process became known as accelerated approval [9].
A surrogate endpoint used for accelerated approval is a marker (e.g., a laboratory measurement, radiographic image, physical sign) that is thought to predict clinical benefit; however, it is not itself a measure of clinical benefit [9]. Likewise, an intermediate clinical endpoint is a measure of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a drug, such as an effect on irreversible morbidity and mortality [9]. The FDA bases its decision whether to accept the proposed surrogate or intermediate clinical endpoint on the scientific support for that endpoint. Studies demonstrating a drug's effect on a surrogate or intermediate clinical endpoint must be "adequate and well controlled," as required by the FD&C Act [28,29].
Using surrogate or intermediate clinical endpoints can save valuable time in the drug-approval process. Of course, the drug company will need to conduct studies to confirm this surrogate endpoint predicts patients will achieve the predicted clinical endpoint, such as increased survival time, through Phase 4 confirmatory trials.
When confirmatory trials verify clinical benefit, the FDA will generally terminate the requirement. Approval of a drug may be withdrawn or the labeled indication of the drug changed if trials fail to verify clinical benefit or do not demonstrate sufficient clinical benefit to justify the drug's associated risks. This could occur if a drug shows a significantly smaller magnitude or duration of benefit than was anticipated based on the observed effect on the surrogate.
FDA's fast track process facilitates the development and expedites review of such drugs that treat serious conditions and fill an unmet medical need. This authority was first created in a 1997 statute.
Determining whether a condition is "serious" is a judgment call. Generally, this decision is based on the drug's ability to impact survival, day-to-day functioning, and/or the likelihood that the condition, if left untreated, will progress from a less severe condition to a more serious one [29]. AIDS, Alzheimer disease, heart failure, and cancer are clear examples of serious conditions.
Drugs are directed at an unmet need if they are being developed to treat or prevent a condition with no current therapy. If there are available therapies, a fast-track drug must demonstrate some advantage, such as:
Showing superior effectiveness and an effect, or improved effect, on serious outcomes
Avoiding serious side effects of an available therapy
Improving the diagnosis of a serious condition when early diagnosis results in an improved outcome
Decreasing a clinically significant toxicity of an available therapy that causes discontinuation of treatment
Addressing an emerging or anticipated public health need
A drug that receives fast-track designation is eligible for some or all of the following:
More frequent meetings with the FDA to discuss the drug's development plan and ensure collection of appropriate data needed to support drug approval
More frequent written communication from the FDA about topics such as proposed clinical trial design and use of biomarkers
Eligibility for accelerated approval and priority review, if relevant criteria are met
Rolling review, whereby a pharmaceutical company can submit completed sections of its biologic license application or NDA for review by the FDA, rather than waiting until the entire NDA is completed and submitted for review
Fast-track designation may be requested by the pharmaceutical company at any time during the drug-development process. The FDA will review the request and make a decision within 60 days based on whether the drug fulfills an unmet medical need for a serious condition.
After a drug receives fast-track designation, early and frequent communication between the FDA and the pharmaceutical company is encouraged throughout the entire drug development and review process. The frequency of communication assures that questions and issues are resolved quickly, often leading to earlier drug approval and access by patients.
The authority to designate a breakthrough product was created in a 2009 statute. A "breakthrough" therapy may sound like a ground-breaking medical discovery or advancement, but only preliminary clinical evidence is necessary to show that a breakthrough drug may substantially improve existing therapies on at least one endpoint. The breakthrough therapy designation aims to speed the development and review of drugs intended to treat a serious or life-threatening disease alone or with other drugs.
It can be difficult to judge whether a drug's improvement over available therapy is substantial. This determination depends on the magnitude of the effect of treatment, such as treatment duration and the observed clinical outcomes. The preliminary clinical evidence should show a clear advantage over available therapy.
Substantial improvement on one or more clinically significant endpoints generally refers to an endpoint measuring an effect on irreversible morbidity and mortality or symptoms from the disease's serious consequences. A clinically significant endpoint can also refer to findings that suggest an effect on irreversible morbidity and mortality or serious symptoms, including:
An effect on an established surrogate endpoint
An effect on a surrogate endpoint or intermediate clinical endpoint considered reasonably likely to predict a clinical benefit (i.e., the accelerated approval standard)
An effect on pharmacodynamic biomarker(s)—an indicator of a drug's effect used to examine links between drug regimens, effects, and tumor response. These markers do not meet the criteria for an acceptable surrogate endpoint but strongly suggest the potential for a clinically meaningful effect on the underlying disease.
A significantly improved safety profile compared with available therapy, such as less dose-limiting toxicity for an oncology agent with evidence of similar efficacy
A drug that receives breakthrough therapy designation is eligible for the following [36]:
All fast-track designation features
Intensive guidance on an efficient drug-development program, starting as early as Phase 1
Organizational commitment involving senior managers
If the drug sponsor requests breakthrough-therapy designation, the FDA can grant the designation after reviewing the submitted data if the agency believes the drug development program may meet the criteria for the designation. The remaining drug-development program can benefit from the designation [15].
Because the breakthrough-therapy designation intends to develop evidence supporting drug approval as efficiently as possible, the FDA does not anticipate that designation requests will be made after submission of the original biologic license application, NDA, or supplement. Designation requests should be received by the FDA before the end of phase of Phase 2 meetings. FDA will respond to these requests within 60 days [15].
Every drug marketed in the United States goes through the FDA's detailed review process. In 1992, however, FDA created a new two-tiered system aimed to improve drug review times: standard review (10 months) and priority review (6 months). A drug can earn priority review designation if it treats a serious or life-threatening condition and would significantly improve the safety or effectiveness of the treatment, diagnosis, or prevention of that condition if approved. Significant improvement may be demonstrated by [30]:
Evidence of increased effectiveness in treatment, prevention, or diagnosis of condition
Elimination or substantial reduction of a drug reaction that may limit treatment
Documented enhancement of patient compliance that is expected to lead to an improvement in serious outcomes
Evidence of safety and effectiveness in a new subpopulation
Although the FDA determines the review designation of every application, an applicant may expressly request priority review [15]. A drug's designation does not affect the length of the clinical trial period nor does it alter the scientific or medical standard for approval or the quality of evidence necessary.
As extensive as CDER's premarket review process is, active post-marketing surveillance of adverse drug effects is also essential. Not all possible side effects of a drug can be anticipated. Clinical trials only involve several hundred to several thousand patients. So, the FDA maintains a system of post-marketing surveillance and risk assessment programs to identify adverse events (e.g., adverse reactions, poisonings) that did not appear during the drug approval process. The agency uses this information to update drug labeling and, on rare occasions, to re-evaluate the approval decision.
The FDA can require manufacturers of certain drugs to conduct post-market studies and clinical biologic products to assess the possible risks associated with the drugs. Under the Food and Drug Modernization Act of 1997, the FDA is required to publish annual reports on the performance of post-market studies and clinical trials that it obliges or requests from manufacturers [31]. These reports provide updates on the post-market requirements or commitments in relation to their original schedule or "milestones," regardless of whether that schedule has been subsequently adjusted. Data on the post-market requirements and commitments public websites are updated quarterly.
The U.S. Food and Drug Administration Amendments Act of 2007 (FDAAA) also requires the FDA to annually review the backlog of post-market commitments relating to safety in order to determine any that should be revised or eliminated, report those determinations to Congress, and assign timeframes for those commitments [33].
The FDAAA also gave the FDA new authorities and responsibilities to enhance drug safety. One of its provisions gave the FDA the authority to require a Risk Evaluation and Mitigation Strategy (REMS) from sponsors to ensure that the benefits of a drug or biologic product outweigh its risks. REMS are required risk management plans that use risk minimization strategies beyond the professional labeling to ensure that the benefits of certain prescription drugs outweigh their risks [33].
The FDA may require a REMS before or after approval if the FDA becomes aware of new safety information and determines REMS is necessary to ensure that the benefits of the drug outweigh the risks. The risk must be a serious risk that is documented in the drug's label. Essentially, a REMS is a safety strategy to manage a known or potential serious risk associated with a medication and to enable patients to have continued access to such medicines by managing their safe use.
Because medications vary widely, each REMS is also different. Elements to assure safe use are required medical interventions or other actions healthcare professionals must execute prior to prescribing or dispensing the drug to the patient. Some actions may also be required in order for the patient to continue on treatment.
The REMS@FDA website (https://www.fda.gov/rems) improves the organization of REMS information online and makes it easier for users to access comprehensive REMS information. Prescribers can search by the name of the REMS, an element of the REMS program, or by an active ingredient with a REMS. In each REMS, there is a section that explains what healthcare providers and pharmacists must know in order to prescribe and/or dispense a drug with a REMS. The site also features links to the prescribing information from the DailyMed site and to the Drugs@FDA information for a product.
The MedWatch program is for health professionals and the public to voluntarily report serious reactions and problems with medical products, such as drugs and medical devices. MedWatch also ensures that new safety information is rapidly communicated to the medical community to improve patient care. All data contained on the MedWatch form will be entered into the FDA Adverse Event Reporting System (FAERS) database. The MedWatch page includes sections on how to report an adverse event, safety information, and publications.
After a drug is approved and marketed, the FDA uses different mechanisms to assure that firms adhere to the terms and conditions of approval described in the application and that the drug is manufactured in a consistent and controlled manner. This is done by periodic, unannounced inspections of drug production and control facilities by the FDA's field investigators and analysts.
Manufacturers of prescription medical products are required by regulation to submit adverse event reports to the FDA. The MedWatch site provides information on mandatory reporting by manufacturers. In addition, drug manufacturers must submit either error and accident reports or drug-quality reports when deviations from current good manufacturing practice regulations occur.
The FDA receives medication error reports on marketed human drugs (including prescription, generic, and over-the-counter drugs) and non-vaccine biologic products and devices. The National Coordinating Council for Medication Error Reporting and Prevention defines a medication error as [37]:
...any preventable event that may cause or lead to inappropriate medication use or patient harm while the medication is in the control of the health care professional, patient, or consumer. Such events may be related to professional practice, health care products, procedures, and systems, including prescribing; order communication; product labeling, packaging, and nomenclature; compounding; dispensing; distribution; administration; education; monitoring; and use.
CDER medication errors program staff reviews medication error reports sent to the Institute for Safe Medication Practices National Medication Errors Reporting Program and MedWatch, evaluates causality, and analyzes the data to provide feedback to others at the FDA.
Adverse events reported to the FDA's MedWatch program provide a vital source of information to the FDA. Today, resources are available to make reporting to MedWatch easier. Anyone can submit a report to MedWatch online at https://www.fda.gov/MedWatch. Reporting forms can be completed online or printed and completed by hand. In addition to reporting as a health professional, patients should be referred to the consumer form.
The FAERS is a computerized information database designed to support the FDA's post-marketing safety surveillance program for all approved drug and therapeutic biologic products. The ultimate goal of FAERS is to improve the public health by providing the best available tools for storing and analyzing safety reports. The reports in FAERS are evaluated by a multidisciplinary staff of safety evaluators, epidemiologists, and other scientists in the CDER Office of Surveillance and Epidemiology to detect safety signals and to monitor drug safety. If a potential safety concern is identified in FAERS, further evaluation is performed, such as conducting studies using other large databases. Based on an evaluation of the potential safety concern, the FDA may take regulatory action to improve product safety and protect the public health, such as requiring changes to the drug's labeling. Drug safety labeling changes are published online.
Reporting of adverse events and medication errors by healthcare professionals and consumers is voluntary in the United States. The FDA may receive these adverse event and medication error reports directly or from the manufacturer who made the drug or biologic product. If a manufacturer receives an adverse event report, they are, in most cases, required to send the report to FDA. The reports received directly from healthcare professionals/consumers and the reports received from manufacturers are both entered into FAERS [34].
A rare disease is defined as one that affects a limited number of people, specifically fewer than 200,000 people in the United States [38]. However, it is important to note that the term can be deceptive. There are approximately 7,000 different rare diseases, collectively affecting as many as 30 million people, or about 10% of the U.S. population. So, while the number of patients with any given rare disease may be small, the cumulative impact on public health is large.
Rare diseases are seen across a wide array of therapeutic areas, including certain types of cancer, urea-cycle disorders, and other genetic disorders, such as Gaucher disease. Rare diseases are chronic, debilitating conditions that are often inherited and frequently lead to early mortality. About half of those affected by rare diseases are children, and many of the diseases lack effective treatments [38].
Under specific circumstances, the FDA will award priority review vouchers to sponsors of rare pediatric disease product applications that meet certain criteria. In addition, a sponsor who receives an approval for a drug or biologic for a "rare pediatric disease" may qualify for a voucher that can be redeemed to receive a priority review of a subsequent marketing application for a different product.
In 2019, the FDA issued a draft guidance titled RarePediatric Disease Priority Review Vouchers: Guidance for Industry. This guidance explains how FDA plans to implement the section of the act that defines a rare pediatric disease. It also describes the process by which sponsors who are interested in receiving rare pediatric disease priority review vouchers may first request designation of their drug or biologic product ("drug") as a drug for a "rare pediatric disease." While such designation is not required to receive a voucher, requesting this in advance will expedite a sponsor's future request for a priority review voucher.
The Orphan Drug Act allows a drug or biologic product that treats a rare disease or condition to be granted special orphan designation or "orphan status" upon request of a sponsor. For a drug to qualify for orphan designation, both the drug and the disease or condition must meet certain criteria specified in the Orphan Drug Act and the FDA's implementing regulations. Orphan designation qualifies the sponsor of the drug for various development incentives of the Orphan Drug Act, including tax credits for qualified clinical testing. A marketing application for a prescription drug product that has received orphan designation is not subject to a prescription drug user fee unless the application includes an indication for other than the rare disease or condition for which the drug was designated.
A sponsor seeking orphan designation for a drug must submit a request for designation to Office of Orphan Products Development with the information required [39]. Each designation request must stand on its own merit. Sponsors requesting designation of the same drug for the same rare disease or condition as a previously designated product must submit their own data and information in support of their designation request. The granting of an orphan designation request does not alter the standard regulatory requirements and process for obtaining marketing approval. Safety and effectiveness of a drug must be established through adequate and well-controlled studies.
The objective of the patient-focused drug development initiative is to gather patient perspectives on the disease severity and current therapeutic options available for certain disease areas. This information is a critical aspect of the FDA's decision making in the drug-review process because it establishes the context in which regulatory decisions are made. The FDA convened with patient stakeholders to host 24 meetings on specific disease areas between 2013 and 2017. Each meeting resulted in a Voice of the Patient Report summarizing the input provided by patients and patient representatives [40].
Some patients may be eligible for use of an investigational medical product outside of a clinical trial. Expanded access is reserved for patients with serious or immediately life-threatening diseases or conditions who have no comparable or satisfactory alternative therapy and who seek access to potentially life-saving investigational drugs [32].
Whenever possible, use of an investigational medical product by a patient as part of a clinical trial is preferable because clinical trials can produce data that may lead to the approval of products and, consequently, make them more widely available. However, when patient enrollment in a clinical trial is not possible (e.g., no ongoing clinical trial is taking place or a patient is not eligible), patients may be able to receive the product, when appropriate, through expanded access.
Under the FD&C Act, a patient may seek individual patient expanded access to investigational products for the diagnosis, monitoring, or treatment of a serious disease or condition if the following conditions are met [41]:
The patient and a licensed physician are both willing to participate.
The patient's physician determines that there is no comparable or satisfactory therapy available to diagnose, monitor, or treat the patient's disease or condition.
The probable risk to the person from the investigational product is not greater than the probable risk from the disease or condition.
The FDA determines that there is sufficient evidence of the safety and effectiveness of the investigational product to support its use in the particular circumstance.
The FDA determines that providing the investigational product will not interfere with the initiation, conduct, or completion of clinical investigations to support marketing approval.
The sponsor (generally the company developing the investigational product for commercial use) or the clinical investigator (or the patient's physician in the case of a single patient expanded access request) submits a clinical protocol (i.e., a document that describes the treatment plan for the patient) that is consistent with the FDA's statute and applicable regulations for INDs or investigational device exemption applications, describing the use of the investigational product.
The patient is unable to obtain the investigational drug under another IND or to participate in a clinical trial.
Also under the FD&C Act statute, a sponsor or a physician may submit a protocol intended to provide widespread access to an investigational product for multiple patients. In this scenario, the FDA will permit the investigational product to be made available under a treatment IND or treatment investigational device exemption if certain criteria are met.
Ensuring patient safety is a priority. The FDA must determine that the potential patient benefit justifies the potential risk of the expanded access use of the investigational drug. The potential risk may not be unreasonable in the context of the disease or condition to be treated. Even with safeguards, there may be significant unknowns about safety and effectiveness.
The rules of expanded access differ slightly for investigational drugs and biologics compared with requirements for all uses. General requirements and criteria that must be met to authorize expanded access, along with listing the requirements for expanded access submissions and describing patient protection safeguards that preserve the ability to develop meaningful data about the use of the investigational product, are provided in 21 CFR part 312 subpart I.
Under the FDA's current regulations for investigational drugs and biologics, there are three categories of expanded access [13]:
Expanded access for individual patients, including for emergency use
Expanded access for intermediate-size patient populations
Expanded access for widespread use
Overall, the FDA authorizes more than 99% of expanded access requests it receives. Treatment may begin 30 days after the FDA receives the IND, or earlier if notified by the FDA. The treating physician must ensure that institutional review board review is obtained in accordance with the FDA's regulations. The treating physician may also be required to provide the IND application number to the medical product company prior to the company shipping the investigational drug. This number will be provided upon FDA authorization of the expanded access request.
To keep healthcare professionals and members of the public abreast of daily updates related to drug approvals, safety, and labeling changes, the FDA offers a variety of email notifications and social media feeds. For example, individuals may follow @FDA_Drug_Info on Twitter for the latest news and information. These communications can help healthcare professionals improve their knowledge of ever-changing FDA-approved medications, which can result in improved patient safety.
MedWatch alerts provide physicians and the public with timely new safety information about human medical products. These alerts can impact treatment and diagnostic choices made by healthcare professionals and their patients. Safety alerts may relate to human drugs, medical devices, vaccines and other biologic products, dietary supplements, and cosmetics.
Physicians can submit a MedWatch Online Voluntary Reporting Form to report problems (e.g., adverse events, medication error reports) related to an FDA-regulated drug. Healthcare professionals may also report these issues to product manufacturers. Manufacturers are required to send any adverse event report to the FDA, as specified by regulations. Reports the FDA receives directly or from manufacturers are entered into FAERS.
The Drug Safety Labeling Changes database informs the public of approved safety labeling changes. Among these labeling changes are updates to boxed warnings, contraindications, warnings, adverse reactions, or patient package insert/medication guide sections of the drug label. Changes to labeling data made before 2016 are available on the MedWatch website.
News about approved drugs and other medical products approved since 2014 are posted online on the press announcements webpage. Subscription to the CDER New email list will result in updates on drug topics, including new and generic drug approvals and drug shortages. Additionally, Drug Trials Snapshots provide clinicians and consumers with information about newly approved drugs and who participates in clinical trials for these drugs.
Since its conception in 1906, the FDA's historical role in drug regulation has evolved to the present-day standards of safety and efficacy for all new drugs. The FD&C Act of 1938 required manufacturers to show that a drug was safe before it could be marketed. Amendments to the Act in 1962 explicitly state that the FDA would rely on scientific testing and that new drug approvals would be based not only upon proof of safety, but also on substantial evidence of a drug's efficacy. By evaluating new drugs before they can be marketed in the United States, CDER helps provide physicians and healthcare professionals with the information they need to use medications wisely.
As the largest of FDA's six centers, CDER helps protect and promote public health by ensuring that drugs marketed in this country are safe and effective. Although the FDA does not test drugs, it analyzes data and proposed labeling in an independent and unbiased review to establish whether a drug's health benefits outweighs any known risks before the drug is approved for sale. The FDA has unique programs, such as rare disease development and orphan drug development, to help apply regulatory flexibility to the heterogeneous nature of disease.
A variety of resources are available for physicians, healthcare professionals, and consumers. From various subscription services to access more information on the latest drug approvals to safety updates, the FDA is continually working to engage with the healthcare professional community to protect the health of all Americans.
1. U.S. Food and Drug Administration. Fact Sheet: FDA at a Glance. Available at https://www.fda.gov/about-fda/fda-basics/fact-sheet-fda-glance. Last accessed July 7, 2023.
2. U.S. Food and Drug Administration. What We Do. Available at https://www.fda.gov/about-fda/what-we-do. Last accessed July 7, 2023.
3. National Center for Health Statistics. Health, United States, 2018. Available at https://www.cdc.gov/nchs/data/hus/hus18.pdf. Last accessed July 7, 2023.
4. Henry J. Kaiser Family Foundation. Number of Retail Prescription Drugs Filled at Pharmacies by Payer. Available at https://www.kff.org/health-costs/state-indicator/total-retail-rx-drugs. Last accessed July 7, 2023.
5. Kesselheim AS, Woloshin S, Eddings W, Franklin JM, Ross KM, Schwartz LM. Physicians' knowledge about FDA approval standards and perceptions of the "breakthrough therapy" designation. JAMA. 2016;315(14):1516-1518.
6. Center for Drug Evaluation and Research. Review Team Responsibilities. Available at https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/review-team-responsibilities. Last accessed July 7, 2023.
7. U.S. Food and Drug Administration. Development and Approval Process: Drugs. Available at https://www.fda.gov/drugs/development-approval-process-drugs. Last accessed July 7, 2023.
8. U.S. Food and Drug Administration. FDA and Clinical Drug Trials: A Short History. Available at https://www.fda.gov/media/110437/download. Last accessed July 7, 2023.
9. U.S. Code of Federal Regulations. Title 21—Food and Drugs, Chapter I—Food and Drug Administration Department of Health and Human Services, Subchapter D—Drugs for Human Use, Part 314—Applications For FDA Approval To Market. Available at https://gov.ecfr.io/cgi-bin/text-idx?SID=fcf70feccc50ec11390054e40a58a5bb&mc=true&node=pt21.5.314&rgn=div5. Last accessed July 7, 2023.
10. U.S. Food and Drug Administration. Guidance Document: Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/providing-clinical-evidence-effectiveness-human-drug-and-biological-products. Last accessed July 7, 2023.
11. Daemmrich A. Pharmacovigilance and the missing denominator: the changing context of pharmaceutical risk mitigation. Pharm Hist. 2007;49(2):61-75.
12. The 87th U.S. Congress. Senate Report No. 1744, Part 2. Available at https://www.govinfo.gov/media/87-2num.pdf. Last accessed July 7, 2023.
13. U.S. Food and Drug Administration. Expanded Access Categories for Drugs (Including Biologics). Available at https://www.fda.gov/news-events/expanded-access/expanded-access-categories-drugs-including-biologics. Last accessed July 7, 2023.
14. U.S. Food and Drug Administration. FDA's Drug Review Process: Continued. Available at https://www.fda.gov/drugs/drug-information-consumers/fdas-drug-review-process-continued. Last accessed July 7, 2023.
15. U.S. Food and Drug Administration. Step 1: Discovery and Development. Available at https://www.fda.gov/patients/drug-development-process/step-1-discovery-and-development. Last accessed July 7, 2023.
16. U.S. Food and Drug Administration. Drug Development and Review Definitions. Available at https://www.fda.gov/drugs/investigational-new-drug-ind-application/drug-development-and-review-definitions. Last accessed July 7, 2023.
17. U.S. Food and Drug Administration. Investigational New Drug (IND) Application. Available at https://www.fda.gov/drugs/types-applications/investigational-new-drug-ind-application. Last accessed July 7, 2023.
18. U.S. Food and Drug Administration. Step 3: Clinical Research. Available at https://www.fda.gov/patients/drug-development-process/step-3-clinical-research. Last accessed July 7, 2023.
19. U.S. Food and Drug Administration. Advisory Committees: Critical to the FDA's Product Review Process. Available at https://www.fda.gov/drugs/drug-information-consumers/advisory-committees-critical-fdas-product-review-process. Last accessed July 7, 2023.
20. Kesselheim AS, Wang B, Franklin JM, Darrow JJ. Trends in utilization of FDA expedited drug development and approval programs, 1987–2014: cohort study. BMJ. 2015;351:h4633.
21. U.S. Food and Drug Administration. Guidance Document: Expedited Programs for Serious Conditions—Drugs and Biologics. Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expedited-programs-serious-conditions-drugs-and-biologics. Last accessed July 7, 2023.
22. U.S. Food and Drug Administration. Novel Drug Approvals for 2019. Available at https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2019. Last accessed June 8, 2020.
23. U.S. Food and Drug Administration. Novel Drug Approvals for 2022. Available at https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2022. Last accessed July 7, 2023.
24. U.S. Food and Drug Administration. Accelerated Approval. Available at https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/accelerated-approval. Last accessed July 7, 2023.
25. Centers for Disease Control and Prevention. Update: mortality attributable to HIV infection among persons aged 25–44 Years—United States, 1991 and 1992. MMWR. 1993;42(45);869-872.
26. 26. U.S. Food and Drug Administration. FDA's Comprehensive Response to HIV. Available at https://www.fda.gov/news-events/fda-voices/fdas-comprehensive-response-hiv-part-i. Last accessed July 7, 2023.
27. U.S. Food and Drug Administration. Food and Drug Administration Safety and Innovation Act (FDASIA). Available at https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/food-and-drug-administration-safety-and-innovation-act-fdasia. Last accessed July 7, 2023.
28. Federal Food, Drug, and Cosmetic Act. Section 355e: Pharmaceutical Security. Available at https://uscode.house.gov/view.xhtml?req=granuleid:USC-prelim-title21-section355e&num=0&edition=prelim. Last accessed July 7, 2023.
29. Federal Food, Drug, and Cosmetic Act. Section 356: Expedited Approval of Drugs for Serious or Life-Threatening Diseases or Conditions. Available at https://uscode.house.gov/view.xhtml?req=granuleid:USC-prelim-title21-section356&num=0&edition= prelim. Last accessed July 7, 2023.
30. U.S. Food and Drug Administration. Priority Review. Available at https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/priority-review. Last accessed July 7, 2023.
31. U.S. Food and Drug Administration. Postmarketing Requirements and Commitments: Reports. Available at https://www.fda.gov/drugs/postmarket-requirements-and-commitments/postmarketing-requirements-and-commitments-reports. Last accessed July 7, 2023.
32. U.S. Food and Drug Administration. Expanded Access. Available at https://www.fda.gov/news-events/public-health-focus/expanded-access. Last accessed July 7, 2023.
33. U.S. Food and Drug Administration. FDA Transparency Initiative Overview. Available at https://www.fda.gov/about-fda/transparency/transparency-initiative. Last accessed June 8, 2020.
34. 34.U.S. Food and Drug Administration. Transcript: FAERS. Available at https://www.fda.gov/drugs/information-healthcare-professionals-drugs/transcript-faers. Last accessed July 7, 2023.
35. Greene JA, Podolsky SH. Reform, regulation, and pharmaceuticals: the Kefauver-Harris Amendments at 50. N Engl J Med. 2012;367(16):1481-1483.
36. U.S. Food and Drug Administration. Frequently Asked Questions: Breakthrough Therapies. Available at https://www.fda.gov/regulatory-information/food-and-drug-administration-safety-and-innovation-act-fdasia/frequently-asked-questions-breakthrough-therapies. Last accessed July 7, 2023.
37. National Coordinating Council for Medication Error Reporting and Prevention. Available at https://www.nccmerp.org/about-medication-errors. Last accessed July 7, 2023.
38. U.S. Food and Drug Administration. Rare Diseases Program. Available at https://www.fda.gov/about-fda/center-drug-evaluation-and-research-cder/rare-diseases-program. Last accessed July 7, 2023.
39. U.S. Food and Drug Administration. Office of Orphan Products Development. Available at https://www.fda.gov/about-fda/office-clinical-policy-and-programs/office-orphan-products-development. Last accessed July 7, 2023.
40. U.S. Food and Drug Administration. CDER Patient-Focused Drug Development. Available at https://www.fda.gov/drugs/development-approval-process-drugs/cder-patient-focused-drug-development. Last accessed July 7, 2023.
41. U.S. Food and Drug Administration. Federal Food, Drug, and Cosmetic Act (FD&C Act). Available at https://www.fda.gov/regulatory-information/laws-enforced-fda/federal-food-drug-and-cosmetic-act-fdc-act. Last accessed July 7, 2023.
42. U.S. Food and Drug Administration. Drug Approval: Bringing a New Drug to the Market. Available at https://www.fda.gov/media/94428/download. Last accessed July 7, 2023.
Mention of commercial products does not indicate endorsement.